Advancing Reproducibility in Solid Electrode Surface Renewal: Strategies for Consistent Electrochemical Interfaces in Biomedical Research
This article provides a comprehensive examination of strategies to enhance the reproducibility of solid electrode surface renewal, a critical process in electroanalysis and biosensing.
Advancing Reproducibility in Solid Electrode Surface Renewal: Strategies for Consistent Electrochemical Interfaces in Biomedical Research
Abstract
This article provides a comprehensive examination of strategies to enhance the reproducibility of solid electrode surface renewal, a critical process in electroanalysis and biosensing. It explores the fundamental mechanisms governing surface regeneration, from electrochemical modification in aqueous media to mechanical renewal techniques. A detailed comparison of methodological approaches, including their optimization for specific biomedical applications like dopamine sensing, is presented. The content further addresses common troubleshooting scenarios and outlines rigorous validation protocols to ensure reliable and consistent electrode performance, ultimately aiming to standardize practices for researchers and professionals in drug development.
Understanding Surface Renewal: Core Principles and Electrode Contamination Challenges
The Critical Impact of Electrode Fouling and Passivation on Analytical Performance
FAQs: Understanding Electrode Fouling and Passivation
What is electrode passivation and how does it differ from fouling? Passivation is the spontaneous formation of a thin, relatively inert film (often an oxide layer) on an electrode surface, which acts as a barrier separating the electrode material from the electrolyte [1] [2] [3]. This phenomenon is central to the operational characteristics of electrochemical energy storage devices, particularly lithium-ion batteries, where the film, known as the Solid Electrolyte Interphase (SEI), is necessary for stability but can be detrimental to ion transfer kinetics [3]. Fouling refers to the undesirable passivation of electrodes, which increases circuit resistance and interferes with electrochemical applications such as amperometric sensing and electrochemical synthesis [1]. Fouling typically occurs through nonspecific modifications of electrode morphology or composition [4].
What are the primary observable consequences of electrode passivation in analytical measurements? The main consequences include decreased rate of electrode reaction resulting in the shift of half-wave potentials, increase in overpotential, distortion of voltammetric waves, decrease in peak currents, and poor reproducibility of analytical signals [5]. These effects collectively undermine analytical performance by reducing sensitivity, increasing detection limits, and compromising measurement precision [6] [5].
Which metals are most prone to spontaneous passivation? Iron, nickel, silicon, chromium, and titanium exhibit significant passivity [1] [2]. Under typical conditions, zinc, cadmium, tin, uranium, and thallium show limited passivity [2]. Aluminum naturally forms a thin surface layer of aluminum oxide on contact with oxygen through oxidation, creating a physical barrier to corrosion [1]. Similarly, titanium immediately forms a titanium dioxide passivation layer when exposed to air, making it resistant to corrosive environments like seawater [1].
What are the mechanisms behind passive film formation? Two primary mechanisms exist: network-forming oxides (Si, Al, Ti) grow by inward O²⁻ diffusion forming dehydrated, compact oxide films without electrolyte anions, while network-modifying oxides (Cu, Fe, Pt) grow by outward metal ion diffusion forming less protective films that include anions [2]. The passivation kinetics can be explained by a two-dimensional phase transition model where isolated adsorbed metal hydroxide ions convert into a condensed metal oxide layer [2].
Troubleshooting Guides: Diagnosis and Resolution
Table 1: Common Electrode Issues and Diagnostic Approaches
| Observed Problem | Potential Causes | Diagnostic Experiments | Immediate Solutions |
|---|---|---|---|
| Current decrease over time | Insulating layer formation (passivation/fouling) [6] [5] | Electrochemical impedance spectroscopy; Surface characterization (XPS, SEM) [6] | Mechanical polishing; Electrochemical cleaning; Ultrasound treatment [5] [7] |
| Potential drift | Buildup of oxide layers or adsorbed species [6] | Cyclic voltammetry in standard solution; Tafel plot analysis [6] [2] | Implement chemical passivation standards (e.g., ASTM A967) [1] |
| Poor reproducibility | Variable electrode surface state between measurements [4] | Statistical analysis of repeated measurements; Surface spectroscopy [5] | Standardized surface renewal protocol; Switch to renewable electrode systems [4] [7] |
| Unexpected reaction products | Side reactions between electrode and solution components [6] | Analysis of solution composition; Characterization of electrode surface [6] | Change electrode material; Modify electrolyte composition [6] |
Table 2: Strategies for Minimizing Electrode Passivation
| Strategy | Principle | Examples | Limitations |
|---|---|---|---|
| Surface Renewal | Physical removal of passivated layer [4] [5] [7] | Mechanical cutting of surface layers (0.1-5 μm) [7]; Sliding metal-coated microwires [4] | Requires human involvement; Complicates automatization [5] |
| Disposable Electrodes | Single-use eliminates cross-contamination [5] | Electrodes from aluminum foil, used CDs, or carbon rods [5] | Increased cost; Not environmentally friendly [5] |
| Surface Modification | Coating prevents fouling [5] | Boron-doped diamond electrodes; Tetrahedral amorphous carbon [5] | Limited robustness and shelf-life [5] |
| Flow Systems | Hydrodynamic removal of passivating species [5] | HPLC-AD; FIA-AD; BIA-AD; Rotating disc electrode [5] | Only effective for reaction products, not matrix components [5] |
Experimental Protocols for Surface Renewability Studies
Protocol 1: Mechanical Surface Renewal of Solid Electrodes
This protocol is adapted from Skvortsova et al. for in situ renewal of electrode surfaces [7].
Materials Required:
- Hexanite-R cutter (serviceability >10⁵ cuts)
- Solid indicator electrodes (Pt, Ag, Au, Cu, Cd, Zn, Co, Ni, or graphite)
- Potentiostat with standard three-electrode configuration
Procedure:
- Mount the electrode in the renewal device ensuring secure positioning
- Align the cutter at optimum angles (specific angles for each electrode material)
- For metallic electrodes: cut 4-5 μm surface layers
- For graphite electrodes: cut 0.1-1.5 μm surface layers
- Perform renewal directly in test solution without air exposure
- Validate renewal quality by measuring electrode surface area reproducibility
Quality Control:
- Surface area reproducibility should achieve RSD = 0.2-0.6% over 10⁴ measurements
- Enhanced electrochemical activity confirmed via standard redox probes
Protocol 2: Renewable Electrodes in Microfluidics
This protocol is adapted from Teixeira et al. for renewable solid electrodes in microfluidic devices [4].
Materials Required:
- PDMS chip with three parallel channels interconnected to one perpendicular top channel
- Metal-coated microwires (working, counter, and pseudoreference electrodes)
- Flow control system (capable of up to 40.0 mL min⁻¹ flow rates)
Procedure:
- Insert microwires into each parallel channel
- Establish flow conditions for specific application
- Perform voltammetric measurements as required
- For renewal: manually pull microwires through channels
- Confirm leakage-free operation after renewal
- Continue measurements with renewed electrode surfaces
Advantages:
- Eliminates electrode cleaning steps
- Enables precise and robust analysis of real samples
- No leakage observed even at high flow rates
Research Reagent Solutions: Essential Materials for Electrode Surface Studies
Table 3: Key Research Reagents and Materials
| Material/Reagent | Function/Application | Key Characteristics |
|---|---|---|
| Hexanite-R Cutter [7] | Mechanical surface renewal of solid electrodes | High durability (>10⁵ cuts); Optimized angles for different materials |
| Metal-coated Microwires [4] | Renewable electrodes in microfluidics | Slide through PDMS channels without leakage; Act as working, counter, and reference electrodes |
| Boron-Doped Diamond (BDD) [5] | Passivation-resistant electrode material | –H terminated surface; High resistance to fouling; Wide potential window |
| Nafion Membrane [8] | Proton-selective membrane in flow batteries | Model system for fouling studies; Sulfonic acid groups for cation affinity |
| Chromate Conversion Coating [1] | Passivation of aluminum alloys | Forms amorphous aluminum chromate coating (0.00001–0.00004 inches thick) |
| Citric Acid Passivation Bath [1] | Alternative to nitric acid for stainless steel | Less toxic and biodegradable; Effective for removing surface iron |
Visualization of Electrode Fouling Mechanisms and Solutions
Electrode Fouling Relationships
Surface Renewal Workflow
In research utilizing solid electrodes, the consistent and reproducible preparation of the electrode surface is not merely a preliminary step—it is a fundamental variable that dictates the success and reliability of subsequent experiments. The surface state controls electron transfer kinetics, dictates the thermodynamics of interfacial processes, and is the stage for all analyte recognition events. This technical support guide is framed within a broader thesis on improving reproducibility in solid electrode surface renewal research. It contrasts two principal methodologies: electrochemical surface modification, which builds functional layers on the electrode, and mechanical renewal, which creates a fresh, pristine surface in situ. Understanding their mechanisms, appropriate applications, and common pitfalls is paramount for researchers in electroanalysis, sensor development, and drug discovery.
What is Electrochemical Surface Modification?
This approach involves altering the chemical composition and properties of an electrode surface through electrochemical processes to impart specific functionality. The goal is not to remove material but to add a designed interface. Common techniques include:
- Electropolymerization: Depositing a conductive polymer film (e.g., polypyrrole, polyaniline) onto the electrode surface.
- Formation of Self-Assembled Monolayers (SAMs): Chemisorbing molecules (e.g., alkanethiols on gold) to create a highly ordered, functional interface.
- Electrodeposition: Depositing metals (e.g., Au nanoparticles) or other materials to enhance surface area or catalytic properties.
- Anodization: Electrochemically growing an oxide layer on valve metals (e.g., Ti, Al).
What is Mechanical Renewal?
Mechanical renewal involves physically abrading the electrode surface in situ (within the electrochemical cell and under electrolyte) to generate a fresh, atomically clean, and reproducible interfacial area. This method, inspired by the classic dropping mercury electrode, is exceptionally effective for eliminating surface contaminants, oxides, and pre-existing films that plague solid electrodes [9]. The process typically uses a rotating disk electrode with an embedded abrasive or a specialized apparatus to polish the surface without breaking the electrochemical circuit.
Key Comparative Data
The table below summarizes the fundamental differences between these two approaches, guiding the initial selection of a methodology.
Table 1: Core Characteristics of Surface Modification and Mechanical Renewal
| Feature | Electrochemical Surface Modification | Mechanical Renewal |
|---|---|---|
| Primary Goal | Introduce new chemical functionality (e.g., selectivity, catalysis). | Generate a pristine, clean, and reproducible base electrode surface. |
| Underlying Mechanism | Electrochemical reactions (oxidation/reduction) leading to film formation or chemical bonding. | Physical abrasion to remove the outer layers of the material. |
| Typical Surface State Outcome | A chemically modified, often polymer-coated or molecularly structured interface. | An atomically fresh, clean metal (or carbon) surface. |
| Key Advantage | Enables tailored sensing and specific interactions; essential for biosensors. | Eliminates history effects and contamination; provides a highly reproducible starting point. |
| Primary Challenge | Reproducibility of the modification process; stability of the layer over time. | Requires specialized equipment for in situ renewal; not all electrode geometries are compatible. |
| Ideal for Studies Of | Affinity-based sensing, catalytic mechanisms, interfacial design. | Fundamental double-layer structure, adsorption thermodynamics, metallic alloy surface segregation [9]. |
Troubleshooting Guides & FAQs
Troubleshooting Electrochemical Surface Modification
Problem: No clear evidence of successful electrode modification after functionalization steps.
- Observation: Cyclic voltammograms (CVs) before and after modification look nearly identical [10].
- Potential Causes & Solutions:
- Incomplete or absent SAM formation: EDC/NHS coupling, for instance, activates carboxyl groups but requires a pre-formed SAM (e.g., a thiolated carboxylic acid) on a gold surface [10].
- Solution: Ensure the foundational modification step (like SAM formation) is performed correctly before subsequent reactions.
- Surface contamination: The presence of contaminants (e.g., silver from reference electrode leakage) can block the sites needed for modification [10].
- Solution: Prior to modification, validate electrode cleanliness by running a CV in a 5 mM potassium ferrocyanide solution. A clean, active surface will show a reversible redox peak. A distorted signal indicates a dirty electrode.
- Incorrect modification chemistry: The chosen reagents or potentials may be unsuitable for the target surface or desired layer.
- Solution: Systematically characterize after each modification step using a redox probe like ferrocyanide. A successful modification should cause the ferrocyanide peaks to decrease and shift due to blocked electron transfer [10].
- Incomplete or absent SAM formation: EDC/NHS coupling, for instance, activates carboxyl groups but requires a pre-formed SAM (e.g., a thiolated carboxylic acid) on a gold surface [10].
Problem: Modified electrode shows poor stability or signal drift during measurement.
- Observation: The baseline drifts or the signal degrades rapidly upon repeated cycling.
- Potential Causes & Solutions:
- Poor adhesion of the modified layer: The film may be delaminating or dissolving.
- Solution: Optimize modification parameters (e.g., scan rate, number of cycles, monomer concentration) to form a more robust film. Consider using cross-linkers.
- Non-specific adsorption: Interfering species from the sample matrix are adsorbing onto the surface.
- Solution: Incorporate blocking agents (e.g., bovine serum albumin) or use more specific recognition elements to passivate non-specific sites.
- Poor adhesion of the modified layer: The film may be delaminating or dissolving.
Troubleshooting Mechanical Renewal
Problem: Unusual peaks appear in the voltammogram after renewal.
- Observation: Symmetrical oxidation/reduction peaks, often around 0 V (vs. Ag/AgCl) [10].
- Potential Causes & Solutions:
- Contamination from the reference electrode: Silver (Ag/AgCl) from the reference electrode can migrate or leak onto the working electrode surface [10].
- Solution: Test a fresh, unused electrode to check for inherent manufacturing defects. If contamination appears after use, avoid harsh electrolytes that dissolve silver and review handling protocols.
- Abrasive particle incorporation: Particles from the renewal process may become embedded in the soft electrode surface.
- Solution: Optimize the renewal pressure and duration. Use abrasives that are chemically inert in the experimental medium.
- Contamination from the reference electrode: Silver (Ag/AgCl) from the reference electrode can migrate or leak onto the working electrode surface [10].
Problem: Irreproducible capacitance or current signals after renewal.
- Observation: The electrical double layer (EDL) capacitance or faradaic current varies significantly between renewal cycles.
- Potential Causes & Solutions:
- Non-uniform surface renewal: The mechanical process may not be creating a geometrically consistent surface area.
- Solution: Standardize the renewal parameters (pressure, speed, time) rigorously. For alloys, be aware that surface segregation—where one component (e.g., Pb in a Sn-Pb alloy) diffuses to the surface over time—can cause the EDL properties to evolve after renewal [9]. The kinetics of this process are solvent-dependent.
- Incomplete renewal: The process may not be removing the entire passivated or contaminated layer.
- Solution: Increase renewal duration or pressure, and verify with a standard redox probe.
- Non-uniform surface renewal: The mechanical process may not be creating a geometrically consistent surface area.
Detailed Experimental Protocols
Protocol: Validating a Clean, Unmodified Electrode Surface
Principle: This is a critical pre-modification check to ensure the base electrode is in a proper state for further functionalization or fundamental studies [10].
- Preparation: Prepare a 5 mM solution of potassium ferrocyanide, K₃[Fe(CN)₆], in a supporting electrolyte like 0.1 M KCl.
- Measurement: Place the electrode (e.g., a cleaned gold or glassy carbon electrode) into the solution and record a cyclic voltammogram (CV) at a moderate scan rate (e.g., 50-100 mV/s).
- Validation: A clean, electrochemically active surface will exhibit a well-defined, reversible redox couple with a peak separation (ΔEp) close to 59 mV for a one-electron transfer process. A large peak separation (>100 mV) or low peak current indicates a contaminated or passivated surface that requires cleaning before use.
Protocol: Stepwise Characterization of Electrode Modification
Principle: To systematically confirm the success of each step in a multi-stage surface modification process [10].
- Step 0 - Baseline: Perform the "Validating a Clean Electrode" protocol (4.1) on the bare electrode. Record the CV and also an Electrochemical Impedance Spectrum (EIS) in the ferrocyanide solution.
- Step 1 - Post-SAM Formation:
- Modify the electrode to form a Self-Assembled Mononlayer (e.g., immerse in a mM solution of a thiol for gold electrodes).
- Rinse thoroughly and repeat the CV and EIS in the ferrocyanide solution.
- Expected Outcome: The redox peaks in the CV will diminish significantly and the charge transfer resistance (Rct) from EIS will increase dramatically, as the SAM blocks electron transfer.
- Step 2 - Post-Ligand Attachment:
- Perform the next modification step (e.g., EDC/NHS coupling to attach a protein or DNA strand).
- Rinse and repeat the electrochemical characterization.
- Expected Outcome: A further increase in Rct and decrease in peak current is often observed, confirming the addition of a new layer.
Protocol: Studying Surface Segregation on Alloys via Mechanical Renewal
Principle: To investigate the kinetics of surface composition changes on a solid alloy electrode after in situ mechanical renewal [9].
- Cell Setup: Use an electrochemical cell equipped with a mechanism for mechanically renewing the solid alloy electrode (e.g., Sn-Pb) in situ under the electrolyte (e.g., LiClO₄ in acetonitrile) [9].
- Initial Measurement: Immediately after renewal, perform a fast capacitance measurement (e.g., via impedance) across a wide potential range to capture the EDL structure of the freshly mixed surface.
- Kinetic Monitoring: Track the evolution of the capacitance-potential (C-E) curves over time at open circuit or a fixed potential. The zero-charge potential (Eσ=0) will shift as the surface composition changes (e.g., from Sn-like to Pb-like for a Sn-Pb alloy) [9].
- Data Analysis: Model the time-dependent coverage (θ) of the surface-active component (e.g., Pb). The process often follows a surface diffusion-controlled mechanism, which is slower in aprotic solvents like acetonitrile compared to water [9].
Workflow and Signaling Pathways
The Scientist's Toolkit: Key Research Reagents & Materials
Table 2: Essential Materials for Surface Renewal and Modification Experiments
| Item Name | Function / Purpose | Example Use Case |
|---|---|---|
| Potassium Ferricyanide [10] | Redox probe for validating electrode cleanliness and activity. | CV in a ferrocyanide solution confirms a clean, unmodified electrode surface is ready for modification or fundamental study. |
| Alkanethiols (e.g., 6-Mercaptohexanoic acid) [10] | Molecules that form Self-Assembled Monolayers (SAMs) on gold surfaces. | Creating a foundational, ordered layer on a gold electrode for subsequent covalent attachment of biomolecules. |
| EDC / NHS Crosslinkers [10] | Carbodiimide chemistry reagents for activating carboxyl groups. | Coupling carboxylic acid-terminated SAMs to amine-containing proteins or DNA strands for biosensor development. |
| Conductive Polymer Precursors (e.g., Pyrrole) | Monomers for electropolymerization to form conductive films. | Electrodepositing a polypyrrole film to enhance charge transfer or to entrap enzymes for catalytic sensing. |
| Lithium Perchlorate (LiClO₄) in Acetonitrile [9] | Aprotic electrolyte for fundamental double-layer studies. | Used in studies of ideal polarizability and surface segregation on renewed metal and alloy electrodes, minimizing solvent interference [9]. |
| Abrasive Disks/Films (e.g., Alumina) | For mechanical polishing and renewal of electrode surfaces. | Creating a fresh, reproducible surface on solid electrodes either ex situ or as part of an in situ renewal apparatus. |
Troubleshooting Guides
Issue: Poor Reproducibility in Electrochemical Measurements
Problem Description Researchers observe inconsistent results between experimental runs when using the same solid electrode, characterized by significant variation in voltammetry peaks or signal drift.
Affected Environments
- All solid electrode types (Pt, Au, Ag, carbon-based)
- Microfluidic electrochemical cells
- Aqueous and non-aqueous electrolytes
Solution Step 1: Verify Electrode Surface Contamination
- Inspect electrode surface under microscope for visible contamination
- Perform cyclic voltammetry in blank electrolyte to check for unexpected redox peaks
- Compare current response with baseline measurements from clean electrode
Step 2: Implement Mechanical Surface Renewal Protocol
- Use a mechanical cutter to remove 0.1-5.0 μm surface layer [7]
- For metallic electrodes (Pt, Ag, Au), cut 4-5 μm layers
- For graphite and graphite-based electrodes, cut 0.1-1.5 μm layers
- Ensure cutter tool angles are optimized for your electrode material
Step 3: Validate Surface Renewal
- Confirm renewed surface area reproducibility (target RSD = 0.2-0.6%)
- Test electrochemical activity using standard redox couple
- Document renewal parameters for future reproducibility
Related Solutions If mechanical renewal is insufficient, consider:
- Electrochemical pre-treatment methods
- Chemical cleaning protocols specific to your electrode material
Issue: Inconsistent Performance of Oxygen-Functionalized Carbon Electrodes
Problem Description Carbon electrodes with introduced oxygen-containing functional groups show varying electrochemical performance, including fluctuating capacitance values or unstable cycling performance.
Affected Environments
- Supercapacitor devices
- Potassium/lithium-ion battery anodes
- Electroanalysis applications
Solution Step 1: Characterize Oxygen Functional Group Composition
- Perform XPS analysis to quantify specific oxygen group types [11]
- Identify ratios of C=O, COOH, C-O-C, and OH groups
- Correlate group composition with electrochemical performance
Step 2: Optimize Functional Group Balance
- Target C=O and COOH groups for enhanced K+ storage performance [11]
- Limit excessive C-O content which promotes undesired SEI components
- Balance oxygen content to maintain electrical conductivity
Step 3: Control Solid Electrolyte Interphase (SEI) Formation
- Monitor SEI composition using in situ FT-IR spectroscopy
- Promote formation of conductive inorganic components (K₂CO₃)
- Minimize resistive organic components (ROCO₂K)
Prevention Tips
- Implement controlled thermal treatment (300-900°C) under H₂/N₂ atmosphere [12]
- Regularly characterize surface chemistry between experimental runs
- Maintain consistent precursor materials and processing conditions
Frequently Asked Questions (FAQs)
Electrode Surface Regeneration
Q: What are the most effective methods for regenerating solid electrode surfaces without compromising surface area reproducibility? A: Multiple approaches show effectiveness depending on application context. Mechanical surface layer cutting (0.1-5 μm) provides excellent reproducibility (RSD 0.2-0.6%) for various electrode materials [7]. For microfluidic applications, manual sliding of metal-coated microwires effectively renews surfaces without treatments, mimicking mercury drop electrode functionality [4]. Thermal treatments under controlled atmospheres (H₂/N₂, 300-900°C) can regenerate carbon electrodes by modifying oxygen functional groups while maintaining pore characteristics [12].
Q: How can I verify that my electrode surface regeneration protocol is successful? A: Success verification should include both physical and electrochemical characterization. For mechanical renewal, measure surface area consistency across multiple cycles (target <0.6% RSD) [7]. Electrochemically, test using standard redox couples to confirm restored activity. For carbon materials, use XPS to verify desired oxygen functional group profiles and electrical impedance spectroscopy to ensure conductivity maintenance [13] [11].
Oxygen-Containing Functional Groups
Q: Which specific oxygen-containing functional groups most beneficially impact electrochemical performance, and which should be minimized? A: Research indicates C=O (carbonyl) and COOH (carboxyl) groups significantly enhance performance in energy storage applications by contributing to capacity through reversible K+ adsorption/desorption and promoting formation of conductive SEI components [11]. Conversely, excessive C-O-C (epoxide) and OH (hydroxyl) content may increase impedance and promote less desirable SEI organic components, potentially compromising long-term stability [11].
Q: What methods allow precise control over the type and content of oxygen functional groups on carbon electrodes? A: Controlled thermal treatment under specific atmospheres provides effective tuning. Heat treatment in hydrogen-containing atmosphere (4% H₂/N₂) at 300-900°C selectively reduces specific oxygen groups while maintaining favorable pore characteristics [12]. Chemical oxidation methods (e.g., Hummers' method) with varying oxidant amounts (KMnO₄) can control introduction levels, followed by systematic characterization using XPS to quantify specific group types [11].
Research Reagent Solutions
Table 1: Essential Materials for Electrode Surface Research
| Item Name | Function/Application | Key Specifications |
|---|---|---|
| Polydimethylsiloxane (PDMS) | Microfluidic chip fabrication for renewable electrode platforms | Elastomeric properties allow wire sliding without leakage [4] |
| Hexanite-R Cutter | Mechanical surface renewal of solid electrodes | Service life >10⁵ cuts; optimized angles for various electrode materials [7] |
| Hydrogen/Nitrogen Mixed Gas | Thermal treatment atmosphere for oxygen group control | 4% H₂/N₂ mixture; heat treatment at 300-900°C [12] |
| (C₂H₅)₄NBF₄/PC Electrolyte | Organic electrolyte for EDLC performance testing | 1 M concentration in propylene carbonate; for coin-cell assembly [12] |
| KMnO₄ Oxidizing Agent | Introduction of oxygen functional groups to carbon materials | Varying amounts (1-5 g) for controlled oxidation levels [11] |
Experimental Workflows & Surface Characterization
Electrode Surface Renewal and Characterization Workflow
Oxygen Functional Group Impact on Electrochemical Performance
Table 2: Performance Characteristics of Surface Renewal Methods
| Renewal Method | Application Scope | Key Parameters | Performance Outcomes | Reproducibility (RSD) |
|---|---|---|---|---|
| Mechanical Cutting [7] | Pt, Ag, Au, Cu, Cd, Zn, Co, Ni, graphite | 4-5 μm layer (metallic)0.1-1.5 μm (graphite) | Restored electrochemical activityIn situ renewal capability | 0.2-0.6%(10⁴ signal measurements) |
| Microfluidic Wire Sliding [4] | Metal-coated microwires in PDMS chips | Manual slidingFlow rates up to 40.0 mL/min | No leakageNo surface treatment required | Comparable to mercury drop electrodes |
| Thermal Treatment [12] | Activated carbon electrodes | 300-900°C in H₂/N₂1 hour duration | Specific capacitance: 62.1 → 81.6 F/gMaintained pore characteristics | Improved cycle life stability |
Table 3: Impact of Specific Oxygen Functional Groups on Carbon Electrodes
| Functional Group Type | Effect on SEI Composition | Impact on Capacity | Role in Conductivity | Recommended Content Strategy |
|---|---|---|---|---|
| C=O (Carbonyl) [11] | Promotes inorganic components (K₂CO₃) | Significant enhancement via reversible K+ adsorption | Moderate impact | Maximize for K+ storage applications |
| COOH (Carboxyl) [11] | Promotes inorganic components (K₂CO₃) | Significant enhancement via reversible K+ adsorption | Moderate impact | Maximize for K+ storage applications |
| C-O-C (Epoxide) [11] | Increases organic components (ROCO₂K) | Limited enhancement | Reduces conductivity | Minimize for optimal performance |
| OH (Hydroxyl) [11] | Increases organic components (ROCO₂K) | Limited enhancement | Reduces conductivity | Minimize for optimal performance |
Exploring 'Self-Renewal' Phenomena in Advanced Catalytic Materials
FAQs: Understanding Self-Renewal in Catalysis
Q1: What is "self-renewal" in the context of catalytic materials? Self-renewal describes a mechanism where a catalyst can regenerate its active surface during operation, sustaining its catalytic activity over extended periods. This phenomenon often involves the continuous exposure of fresh active sites, counteracting typical deactivation pathways. For instance, in a specific Fe–N–C catalyst, a unique self-renewal mechanism involving layer-by-layer shedding of an iron polyphthalocyanine (FePPc) shell was observed. This shedding process exposes fresh active sites to the electrolyte, which helps maintain the initial catalytic activity for the oxygen reduction reaction (ORR) [14].
Q2: Why is research on self-renewal critical for improving experimental reproducibility? Research into self-renewal is intrinsically linked to reproducibility because it addresses one of the most significant sources of variability in electrochemistry: the unstable and dynamically changing electrode surface. A catalyst with self-renewing properties can maintain a more consistent and well-defined surface state over time and across different experimental setups. This consistency is a fundamental prerequisite for obtaining reproducible performance data, a challenge highlighted by large interlaboratory studies in related fields like all-solid-state batteries [15]. Understanding and controlling self-renewal mechanisms can thus lead to more reliable and comparable research outcomes.
Q3: What are the common failure modes for catalysts that lack self-renewal capabilities? Catalysts without effective self-renewal mechanisms are prone to several deactivation pathways, including:
- Coking: The formation and deposition of carbonaceous materials that block active sites and pores [16].
- Poisoning: The strong chemisorption of species from the feed stream onto active sites [16].
- Sintering: The thermal degradation where supported metal particles agglomerate, reducing the active surface area [16].
- Demetallation/Dissolution: The loss of active metal centers from the catalyst structure into the solution, a common issue for molecular catalysts like iron phthalocyanine in acidic environments [14].
Q4: What advanced characterization techniques are essential for studying self-renewal? To conclusively identify and study a self-renewal process, a combination of techniques is required:
- In situ/operando methodologies: Techniques like Raman spectroscopy and X-ray photoelectron spectroscopy allow for the real-time observation of the catalyst's surface chemistry and structure during operation [17].
- Electrochemical methods: Chronoamperometric stability tests and cyclic voltammetry are used to track performance decay and regeneration over time [14].
- Microscopy: Scanning and transmission electron microscopy (SEM/TEM) are crucial for observing morphological changes, such as the shedding of material layers [17] [14].
- Theoretical calculations: Density functional theory (DFT) helps elucidate the interaction energies and electron transfer processes that underpin the self-renewal mechanism [14].
Troubleshooting Guide: Common Experimental Challenges
The table below summarizes specific issues, their diagnostic data, and solutions related to working with and reproducing self-renewal catalytic systems.
| Problem Observed | Possible Cause | Diagnostic Data to Collect | Proposed Solution |
|---|---|---|---|
| Irreversible activity decay | Leached metal ions aggregating into inactive clusters instead of regenerating active sites [14]. | Inductively Coupled Plasma (ICP) analysis of electrolyte post-testing; TEM for spent catalyst. | Strengthen metal-site anchoring via covalent integration into polymeric structures (e.g., FePPc) rather than simple adsorption [14]. |
| Poor batch-to-batch reproducibility of catalyst synthesis | Inconsistent polymerization or anchoring of molecular precursors to the carbon support. | XPS to compare surface atomic concentrations (Fe, N, C) between batches; reproducibility of half-wave potential (E1/2) [14]. | Adopt controlled synthesis like microwave-assisted polymerization for uniform shell formation [14]. Implement strict precursor quality control. |
| High and drifting background potential in potentiometry | Unstable solid-contact layer in all-solid-state electrodes, leading to ill-defined interfacial potentials and "parallel drift" [18]. | Open Circuit Potential (OCP) measurement over 24+ hours; potentiometric calibration curve slope deviation [18]. | Use hydrophobic, high-capacitance solid-contact materials (e.g., 3D mesoporous carbon) and standardize a 24-hour conditioning protocol [18]. |
| Low catalyst mass loading | Inefficient anchoring of molecular active sites onto the substrate. | Measure Fe mass loading via elemental analysis; compare to theoretical values. Target loadings >2.0 wt% for Fe-N-C systems [14]. | Employ in situ polymerization to build a polymeric shell on the substrate, enabling higher active site density versus molecule adsorption [14]. |
| Inconsistent electrode performance in press cells | Variable microstructure due to uncontrolled pressure application during cell assembly [15]. | Record thickness pre/post pressing; report applied pressures (MPa) and duration for each compression step [15]. | Standardize and meticulously document all assembly parameters, especially stack pressure. Report data in triplicate [15]. |
Key performance metrics and synthesis parameters from seminal research on self-renewing catalysts are summarized in the table below for easy comparison and benchmarking.
| Metric | FePPc/CNT Catalyst (Self-Renewal) | Conventional FePc/CNT Catalyst | Measurement Context |
|---|---|---|---|
| Fe Mass Loading | 2.92 wt% [14] | 0.80 wt% [14] | Material Synthesis |
| Half-wave Potential (E1/2) | 0.74 V vs. RHE [14] | Not specified, but lower activity implied | ORR Activity in 0.1 M HClO4 |
| Tafel Slope | 51 mV dec-1 [14] | Not specified | ORR Kinetics |
| Stability (Current Retention) | ~80% after 24 hours [14] | 42% after 5 hours [14] | Chronoamperometric Test |
| Critical Synthesis Parameter | Microwave-assisted in situ polymerization [14] | Direct adsorption from solution [14] | Synthesis Method |
Experimental Protocol: Synthesis and Testing of a Self-Renewal Fe–N–C Catalyst
Objective: To synthesize an iron polyphthalocyanine shell on carbon nanotubes (FePPc/CNT) and evaluate its self-renewal behavior during the acidic oxygen reduction reaction (ORR) [14].
Part A: Catalyst Synthesis
CNT Support Purification:
- Thermally treat commercial multi-walled CNTs at 450°C in air for 2 hours to remove amorphous carbon.
- Dispense the resulting material in a 3 M HCl aqueous solution. Sonicate for 1 hour, then stir for 12 hours.
- Recover the CNTs via vacuum filtration and wash with deionized water until the filtrate reaches a neutral pH.
- Dry the CNTs in a vacuum oven at 80°C and subsequently anneal them at 1200°C for 3 hours under a 5% H2/Ar flow.
Microwave-Assisted Polymerization of FePPc Shell:
- Disperse 20 mg of the purified CNTs in 10 mL of anhydrous DMF via bath sonication for 1 hour.
- In a separate vial, dissolve 20 mg of 1,2,4,5-tetracyanobenzene (TCNB) and 17.1 mg of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in 10 mL of DMF.
- Combine the two mixtures and add 10.4 mg of anhydrous FeCl2. Sonicate the final mixture for 1 hour.
- Transfer the solution to a microwave reactor and heat at 120°C for 4 hours.
- Once cooled, recover the solid product (FePPc/CNT) by vacuum filtration. Wash sequentially with 1 M HCl, DMF, ethanol, and copious amounts of DI water.
- Dry the final catalyst in a vacuum oven at 80°C for 24 hours.
Part B: Electrochemical Evaluation of Activity and Stability
Ink Preparation and Electrode Fabrication:
- Prepare an ink by dispersing 5 mg of the FePPc/CNT catalyst in a solution containing 950 µL of isopropyl alcohol and 50 µL of 5% Nafion solution. Sonicate for at least 30 minutes to form a homogeneous suspension.
- Pipette a precise volume (e.g., 10-20 µL) of the ink onto a polished glassy carbon rotating disk electrode (RDE) and allow it to dry at room temperature, resulting in a uniform catalyst film.
ORR Activity Measurement:
- Use a standard three-electrode electrochemical cell with the catalyst-coated RDE as the working electrode, a Pt wire as the counter electrode, and a reversible hydrogen electrode (RHE) in the same electrolyte as the reference electrode.
- Fill the cell with 0.1 M HClO4 electrolyte. Saturate the electrolyte with O2 gas.
- Record cyclic voltammograms (CVs) in an O2-saturated atmosphere and linear sweep voltammetry (LSV) curves at a rotation rate of 1600 rpm to obtain the ORR polarization profile. Extract the half-wave potential (E1/2) from the LSV data.
Stability Test to Probe Self-Renewal:
- Perform a chronoamperometric test by holding the catalyst electrode at a constant potential (e.g., 0.4 V vs. RHE) in O2-saturated 0.1 M HClO4 for 24 hours.
- Monitor the current density over time. A stable or slowly decaying current, followed by potential recovery in subsequent CVs, can indicate a self-renewal process where active sites are regenerated.
Part C: Post-Mortem Analysis for Self-Renewal Evidence
- TEM Analysis: Characterize the catalyst morphology after stability testing. Look for evidence of the thinning of the FePPc shell and the presence of shed fragments or sedimented nanoclusters on the CNT support [14].
- ICP-MS Analysis: Analyze the electrolyte after stability testing to quantify the amount of leached iron ions, which is part of the deactivation pathway alongside self-renewal [14].
Visualizing Self-Renewal and Deactivation Pathways
The following diagram illustrates the competing processes of self-renewal and irreversible deactivation in a polymeric shell catalyst system.
Catalyst Lifecycle Pathways
The Scientist's Toolkit: Research Reagent Solutions
The table below lists essential materials and their functions for synthesizing and characterizing self-renewal catalytic systems.
| Reagent/Material | Function in Research | Specific Example / Rationale |
|---|---|---|
| 1,2,4,5-Tetracyanobenzene (TCNB) | Monomer precursor for building the polyphthalocyanine (PPc) polymeric network [14]. | Serves as the molecular building block that, with a metal source, forms the FePPc shell on the CNT support. |
| Multi-walled Carbon Nanotubes (CNTs) | Conductive support substrate [14]. | Provides a high-surface-area, electron-conducting path. Purification (acid washing/annealing) is critical for reproducible performance. |
| Iron(II) Chloride (FeCl2) | Metal source for creating the Fe–N4 catalytic active sites [14]. | Incorporated during polymerization to form the single-atom Fe–N4 centers within the PPc matrix. |
| 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) | Base catalyst for the cyclotetramerization reaction during polymerization [14]. | Promotes the formation of the phthalocyanine rings from TCNB monomers. |
| Perchloric Acid (HClO4) | Electrolyte for acidic ORR testing [14]. | Provides a standard, harsh acidic environment (pH ~1) to rigorously test catalyst stability and self-renewal behavior. |
| Lithium Sulfide (Li2S) & Phosphorus Pentasulfide (P2S5) | Precursors for solid-state electrolyte synthesis [15]. | Used in benchmarking studies to create Li6PS5Cl argyrodite solid electrolyte for all-solid-state battery cells. |
Proven Protocols: Electrochemical and Mechanical Renewal Techniques for Solid Electrodes
This guide provides a detailed protocol for the electrochemical activation of carbon fiber microelectrodes (CFMs) in deionized water. Proper activation is a critical pretreatment step that enhances electrode sensitivity and reproducibility by introducing surface functional groups and increasing the electroactive area. This guide is structured within a broader research context focused on improving the reproducibility of solid electrode surface renewal studies, a common challenge in electrochemical sensing and drug development [19].
Standard Operating Procedure: Electrochemical Activation
Materials and Equipment
| Item | Specification | Purpose/Function |
|---|---|---|
| Carbon Fiber Microelectrode | 7-10 µm diameter | The working electrode whose surface is to be activated and functionalized. |
| Counter Electrode | Platinum wire or mesh | Completes the electrical circuit for current flow. |
| Reference Electrode | Ag/AgCl (or similar) | Provides a stable, known potential for the working electrode. |
| Electrolyte Solution | High-purity deionized water (>18 MΩ·cm) | Medium for electrochemical activation; purity is critical to prevent contamination [20]. |
| Potentiostat/Galvanostat | -- | Instrument to apply controlled potentials/currents and measure electrochemical response. |
Step-by-Step Activation Protocol
Initial Electrode Inspection and Cleaning: Visually inspect the carbon fiber under a microscope for any visible damage or contamination. If necessary, rinse gently with pure ethanol and deionized water.
Electrochemical Cell Setup: Place the carbon fiber microelectrode, counter electrode, and reference electrode into a clean electrochemical cell containing deionized water. Ensure electrodes are properly spaced and not touching.
Application of Activation Signal: Connect the electrodes to the potentiostat. Apply a cyclic voltammetry (CV) waveform with the following typical parameters [21]:
- Potential Window: 0.0 V to +1.5 V (vs. Ag/AgCl)
- Scan Rate: 100 mV/s
- Number of Cycles: 20-30 cycles
Process Monitoring: During the CV cycles, you should observe a steady increase in the background charging current. This indicates successful etching of the carbon surface and an increase in the electroactive area.
Post-Activation Rinsing and Storage: After the final cycle, remove the electrode from the cell and rinse it thoroughly with deionized water to remove any loose surface species. Store the activated CFM in a clean, dry environment if not used immediately.
Troubleshooting Common Experimental Issues
Problem: Low or Unchanging Background Current During Activation
- Potential Cause 1: Electrode Passivation. An insulating film or contamination on the carbon fiber surface is blocking the electrochemical reaction [21].
- Solution: Gently polish the electrode tip with fine alumina slurry (0.05 µm) and sonicate in deionized water for 1-2 minutes to remove the film before reactivation.
- Potential Cause 2: Incorrect Electrical Connections or Cell Setup.
- Solution: Verify all cables are secure, the potentiostat is properly configured, and that no air bubbles are trapped on the electrode surface.
Problem: High Background Noise or Unstable Current
- Potential Cause 1: Contaminated Electrolyte. Impurities in the deionized water can cause parasitic reactions and noise [20].
- Solution: Use fresh, high-purity deionized water (resistivity >18 MΩ·cm). Ensure the electrochemical cell and electrodes are meticulously cleaned.
- Potential Cause 2: Loose Electrical Connections.
- Solution: Check and secure all connections. Ensure the reference electrode is stable and functioning correctly.
Problem: Physical Degradation or Damage to the Carbon Fiber
- Potential Cause: Excessively Harsh Activation Parameters. Applying too high a potential or too many cycles can etch and weaken the fiber [21].
- Solution: Optimize the activation protocol. Reduce the upper potential limit or the number of cycles. Always start with the mildest effective parameters.
Frequently Asked Questions (FAQs)
Q1: Why is deionized water used instead of a traditional acidic or basic electrolyte? Deionized water minimizes the introduction of exogenous ions that can adsorb to the carbon surface and interfere with subsequent experiments, especially in biological sensing. It allows for a cleaner activation process that primarily generates oxygen-containing functional groups from the water itself.
Q2: How can I quantitatively confirm that my activation was successful?
Successful activation is confirmed by both a qualitative and quantitative increase in electrochemical activity. Compare the cyclic voltammograms of a standard redox probe (e.g., 1 mM Ferricyanide, [Fe(CN)₆]³⁻/⁴⁻) before and after activation. A significant decrease in the peak-to-peak separation (ΔEp) and an increase in peak current indicate improved electron transfer kinetics and a larger electroactive area.
Q3: My activated electrodes have poor reproducibility between batches. What could be wrong? Inconsistent electrode performance often stems from variability in pretreatment or surface contamination [19]. Implement a strict, standardized cleaning protocol before activation. Ensure all solutions are prepared consistently, and environmental factors (e.g., temperature) are controlled. Using a structured framework like DMAIC (Define, Measure, Analyze, Improve, Control) can help identify and control these sources of variation [19].
Q4: How long do activated carbon fiber microelectrodes remain stable? The stability can vary from hours to several days, depending on storage conditions and the application. Surface functional groups can slowly reorganize or absorb contaminants. For best results, use the electrodes immediately after activation and validate their performance with a standard probe prior to each critical experiment.
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function in Research |
|---|---|
| Standard Redox Probes (e.g., Potassium Ferricyanide, Dopamine) | Used to quantitatively characterize electrode performance (sensitivity, kinetics) before and after surface renewal/activation. |
| High-Purity Solvents & Salts (e.g., DI Water, KCl, Phosphate Buffers) | Form the electrolyte solution; purity is paramount to prevent surface contamination and unwanted side reactions [20]. |
| Polishing Supplies (e.g., Alumina Slurries, Micropolishing Cloths) | For mechanical surface renewal and removal of old layers or passivating films to restore a baseline surface condition [22]. |
| Surface Characterization Tools (e.g., Raman Spectrometry, SEM) | Used to correlate electrochemical performance with physical/chemical surface changes (e.g., defect density, morphology) from activation [19]. |
Experimental Workflow and Logical Relationships
The following diagram illustrates the key decision points and processes in the electrode activation and troubleshooting workflow.
Key Performance Metrics and Target Values
The table below summarizes the key quantitative metrics to evaluate activation success and the target values to aim for.
| Performance Metric | Method of Measurement | Target Value / Benchmark for Success |
|---|---|---|
| Increase in Electroactive Area | Calculating from CV of 1 mM [Fe(CN)₆]³⁻/⁴⁻ using Randles-Sevcik equation |
>50% increase relative to pre-activation area |
| Improvement in Electron Transfer Kinetics | Peak-to-peak separation (ΔEp) in CV of 1 mM [Fe(CN)₆]³⁻/⁴⁻ |
ΔEp < 80 mV (for a reversible system) |
| Background Charging Current | Charging current at a set potential in CV (e.g., +0.5 V) in DI water | Stable, sigmoidal-shaped increase over activation cycles |
| Surface Functional Group Density | Raman Spectroscopy (ID/IG ratio) [19] | ID/IG ratio optimized for specific application (indicates defect density) |
Mechanical surface renewal is a targeted engineering strategy designed to maintain consistent, reproducible surface conditions during experimental processes. It involves the periodic or continuous physical removal of accumulated deposits, reaction products, or boundary layers from a solid surface. Within the context of solid electrode research, this technique is crucial for sustaining consistent mass transfer rates and active surface area, thereby directly addressing key sources of experimental variability. The core principle hinges on mechanically disrupting the diffusion boundary layer and eliminating porous product layers that impede further reaction, ensuring that the surface remains in a known, reproducible state throughout an experiment [22].
The significance for reproducibility in solid electrode studies cannot be overstated. Many electrochemical reactions, including cementation and other deposition processes, are diffusion-controlled [22]. Over time, the formation of porous solid deposits on the electrode surface acts as a physical barrier, progressively slowing the reaction rate and altering the system's kinetics. Furthermore, the solution at the electrode-solution interface becomes depleted of reactants, creating a concentration gradient. Without intervention, these factors introduce significant time-dependent variables. Mechanical surface renewal counteracts this by periodically re-exposing fresh electrode material and replacing the depleted interfacial solution with fresh bulk solution, maintaining a consistent reaction environment essential for obtaining reproducible, comparable data [22]. The challenge of reproducibility is a noted concern in other fields involving complex surface phenomena, underscoring the need for controlled methodologies [23].
Experimental Protocols: Wiper-Based Surface Renewal
The following detailed methodology is adapted from a seminal study on a wiper-assisted cementation reactor, providing a template for implementing mechanical renewal in electrode systems [22].
Apparatus Setup and Workflow
The foundational setup involves a cylindrical batch reactor where the solid electrode (e.g., a zinc sheet) lines the inner wall. A key innovation is a rotating U-shaped wiper, constructed from plastic-coated steel rods, which simultaneously agitates the bulk solution and mechanically renews the electrode surface.
Diagram of the core experimental workflow for a wiper-based surface renewal system.
Step-by-Step Procedure
- Electrode Surface Pre-treatment: Prior to each experimental run, the electrode surface (e.g., a zinc cylinder) must be meticulously prepared. This involves washing with a 10% HCl solution to remove any passive oxide films, followed by thorough rinsing with distilled water to eliminate acid residues, and finally drying [22].
- Solution Preparation: Prepare the reactant solution (e.g., CuSO₄ solution for cementation studies) using analytical grade chemicals and distilled water. The initial concentration (
C₀) must be accurately known [22]. - Reactor Assembly: Line the inner wall of the cylindrical reactor with the pre-treated electrode sheet. Ensure the back of the electrode is insulated (e.g., with epoxy resin) to confine reactions to the defined active surface. Install the U-shaped wiper mechanism, ensuring it is set to just touch the electrode surface without creating significant air gaps. The wiper is connected to a variable-speed digital motor [22].
- Process Initiation and Sampling: Add a known volume of the prepared solution to the reactor. Start the wiper rotation at the predetermined speed and begin timing. Withdraw small, fixed-volume samples (e.g., 5 mL) from the bulk solution at consistent time intervals throughout the experiment [22].
- Sample Analysis and Data Processing: Analyze the concentration (
C) of the reactant in each sample using an appropriate analytical technique, such as UV-Vis spectrophotometry. The percentage removal of the reactant at timetis calculated as:% Removal = [(C₀ - C)/C₀] × 100[22]. For mass-transfer controlled reactions, the mass transfer coefficient (k) is a critical parameter for comparing system performance under different conditions. It is determined from the slope of a plot ofln(C₀/C)versus time (t), derived from the integrated batch reactor equation:ln(C₀/C) = (k A / Q) t, whereAis the active electrode area andQis the solution volume [22].
Operational Parameter Optimization
The efficiency of mechanical surface renewal is governed by several operational parameters. The data below, derived from a model system, illustrates their quantitative impact.
Table 1: Effect of Operational Parameters on Renewal Efficiency [22]
| Parameter | Variable Range Tested | Observed Effect on Cementation Rate / Mass Transfer Coefficient | Recommended Optimization Strategy |
|---|---|---|---|
| Rotational Speed | Varied RPM | Increased significantly with higher RPM. | Increase speed to enhance renewal frequency and turbulence, but balance against energy consumption and potential for surface damage. |
| Wiper Diameter | Different diameters | Increased with larger diameter wipers. | Use a larger diameter wiper to improve bulk agitation and surface scraping efficiency. |
| Solution pH | Various pH levels | No significant effect, confirming mass-transfer control. | Prioritize control of other parameters; pH can be set based on other experimental needs (e.g., solubility). |
| Initial Concentration | Different Cu²⁺ concentrations | Increased with higher initial concentration. | Use relevant concentration for the study, as it directly drives the concentration gradient. |
Troubleshooting Guide: Common Issues and Solutions
Table 2: Troubleshooting Common Problems in Mechanical Surface Renewal Systems
| Problem | Potential Causes | Diagnostic Steps | Corrective Actions |
|---|---|---|---|
| Inconsistent or Declining Reaction Rate | 1. Wiper not making uniform contact with the surface.2. Inadequate wiper rotational speed.3. Wear and tear on the wiper edges. | 1. Visually inspect the wiper alignment and contact.2. Measure the rate constant over time; a steady decline suggests poor renewal.3. Check wiper for physical damage. | 1. Readjust wiper to ensure it just touches the surface uniformly along its path.2. Increase the wiper's rotational speed within optimal limits.3. Replace or refurbish the wiper. |
| Excessive Vibration or Noise | 1. Unbalanced wiper assembly.2. Spindle or bearing wear.3. Resonant frequency (chatter) from self-excited vibration. | 1. Listen for changes in sound with speed variation.2. Perform a "tap test" to identify loose components.3. Check for patterns on the surface that indicate chatter [24]. | 1. Rebalance the wiper mechanism.2. Tighten all fittings and check/replace bearings.3. Increase machine stability: Increase stiffness of components, or lower process stiffness by reducing workpiece/wheel speed or contact width [24]. |
| Non-Uniform Deposit Removal | 1. Uneven surface of the base electrode.2. Flexible wiper material deforming under load.3. Runout in the wiper drive shaft. | 1. Inspect the electrode surface for flatness/roundness.2. Observe wiper operation under load.3. Use a dial indicator to measure shaft runout. | 1. Machine or polish the electrode to achieve a uniform surface.2. Use a more rigid material for the wiper.3. Correct shaft alignment or lapping to eliminate runout [24]. |
| High Power Consumption | 1. Excessive rotational speed.2. Too much friction between wiper and surface.3. High viscosity solution. | 1. Monitor power with a wattmeter at different speeds [22].2. Check for signs of excessive wear on both wiper and surface. | 1. Optimize speed; use a larger diameter wiper at a lower speed for energy efficiency [22].2. Ensure contact is minimal yet effective.3. Consider operating temperature to modulate viscosity. |
Frequently Asked Questions (FAQs)
Q1: How does mechanical surface renewal directly improve experimental reproducibility? It addresses two primary sources of variability: the buildup of porous solid deposits that physically block the active surface and alter reaction kinetics, and the development of a diffusion boundary layer where reactant concentration is depleted. By periodically scraping the surface, renewal maintains a consistent, known active surface area and a steep concentration gradient, leading to more consistent and reproducible reaction rates over time [22].
Q2: My reaction is not mass-transfer controlled. Is mechanical surface renewal still beneficial? The primary and most quantifiable benefit of mechanical renewal is in mass-transfer controlled systems. If your reaction is kinetically controlled, the benefits may be less pronounced. However, it can still be useful for maintaining a clean and consistent electrode surface, free from fouling or passivation layers that could introduce variability, thus improving reproducibility even in some kinetically limited systems.
Q3: What are the key considerations when selecting a wiper material? The wiper material must be chemically inert to the solution to avoid contamination or corrosion. It should have sufficient mechanical strength and rigidity to perform the scraping action without significant deformation. Furthermore, its hardness should be selected to effectively remove deposits without causing excessive wear to the underlying electrode material. A common approach is to use a rigid core (e.g., steel) coated with an inert polymer [22].
Q4: Can the "surface renewal" concept be applied in other scientific contexts? Yes, the surface renewal model is a well-established principle in micrometeorology for measuring the exchange of heat, water vapor, and gases between the earth's surface and the atmosphere. In this context, it analyzes high-frequency temperature data to estimate sensible heat flux, which is calibrated against eddy covariance measurements [25] [26]. The underlying physical concept of replacing a "stale" surface layer with a fresh one is universal.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Materials and Reagents for Wiper-Based Surface Renewal Experiments
| Item | Example / Specification | Function in the Experiment |
|---|---|---|
| Electrode Material | Zinc, other metal sheets or foils. | Acts as the solid substrate where the reaction (e.g., cementation) and deposit formation occur. Its surface is the subject of renewal. |
| Wiper Mechanism | U-shaped rods, plastic-coated steel. | The core renewal component. It mechanically scrapes the electrode surface to remove deposits and agitates the bulk solution. |
| Reactant Salt | CuSO₄•5H₂O, Analytical Reagent (A.R.) Grade. | Provides the metal ions (e.g., Cu²⁺) for the deposition reaction. Purity is critical for reproducible solution concentration. |
| Cleaning Solution | 10% Hydrochloric Acid (HCl). | Used for pre-treatment etching of the electrode surface to remove oxide layers and ensure a consistent, clean starting state [22]. |
| Spectrophotometry Kit | UV-Vis Spectrophotometer, Cuvettes, standards for calibration. | For quantitative analysis of reactant concentration in sampled aliquots over time, enabling kinetic analysis [22]. |
| Data Acquisition | Variable-speed motor, wattmeter. | To precisely control the wiper RPM and measure the associated energy consumption of the renewal process [22]. |
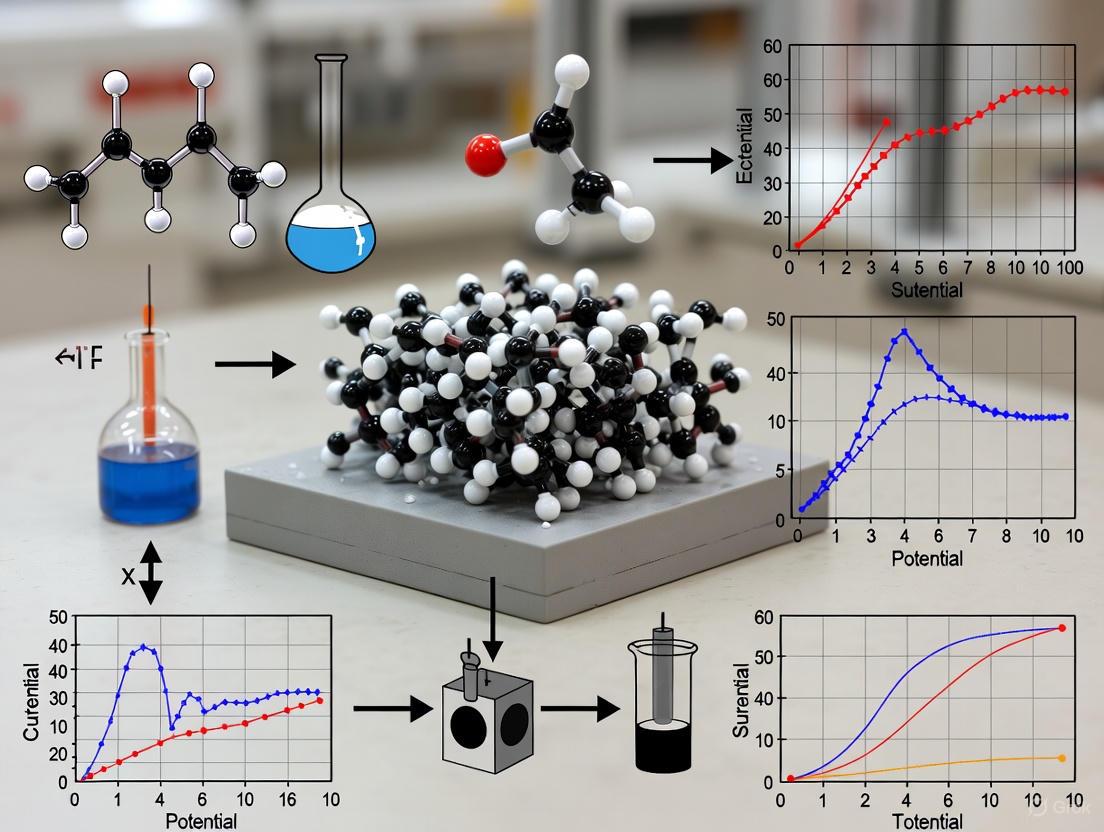
Reproducibility in electrochemical sensing, particularly for neurotransmitters like dopamine, is fundamentally linked to the consistent renewal of solid electrode surfaces. Dopamine is an electroactive catecholamine neurotransmitter crucial for cognitive and behavioral functions, and its imbalance is associated with disorders like Parkinson's disease, schizophrenia, and depression [27]. Electrochemical detection leverages the 2-electron/2-proton redox reaction of dopamine, but faces challenges including electrode fouling from polymerization byproducts and interference from compounds like ascorbic acid (AA) and uric acid (UA) which oxidize at similar potentials [27] [28]. A standardized electrode renewal protocol is therefore essential to ensure that sensing data reflects true analyte concentration rather than inconsistent electrode history.
Key Research Reagent Solutions for Dopamine Sensing
The selection of electrode materials is critical for developing sensitive and selective dopamine sensors. The table below summarizes key materials documented in recent literature.
Table 1: Key Electrode Materials and Reagents for Dopamine Sensing
| Material/Reagent | Function/Benefit | Reported Performance in Recent Studies |
|---|---|---|
| Europium-doped CaZrO3 (ECZO) NPs | Enhances electron transfer and catalytic activity; used in modified carbon paste electrodes (MCPE). | LOD: 0.455 µM; Stability: 92% after 20 cycles; Sensitivity: Peak current of 78.9 µA [29]. |
| Poly(ANSA)/GCE | Electropolymerized film on glassy carbon electrode; increases electroactive surface area. | LOD: 0.089 µM; Linear Range: 0.5 – 100 µM; Diffusion Coefficient: 8.7 × 10⁻⁶ cm²s⁻¹ [30]. |
| NiO/ZnO Hybrid Material | Green-synthesized using date fruit extract; provides high electrocatalytic activity for non-enzymatic sensing. | LOD: 0.036 µM; Exhibits excellent repeatability, selectivity, and reproducibility [31]. |
| PEDOT-PPy Hybrid | Conducting polymer composite on GCE; offers high conductivity, stability, and a large surface area. | LOD: 5 nM; Linear Range: 5 nM to 200 µM; Sensitivity: 7.27 µA/µM cm² [28]. |
| Carbon Nanotubes (CNTs) | Provide high surface area and excellent electrocatalytic properties; often used as a base nanomaterial in composites. | Multi-walled CNTs (MWCNTs) are particularly noted for better performance due to higher surface area for dopamine interaction [32]. |
Experimental Protocols for Electrode Modification and Sensing
Detailed and consistent experimental protocols are the cornerstone of reproducible research. Below are methodologies for key electrode modifications cited in this guide.
- Objective: To create a highly sensitive and selective dopamine sensor via electropolymerization.
- Materials: Glassy Carbon Electrode (GCE), 8-amino-naphthalene-2-sulfonic acid (ANSA), acidic solution (e.g., 0.1 M H₂SO₄), dopamine standard solution, phosphate buffer saline (PBS, pH 7.3).
- Step-by-Step Procedure:
- Electrode Pre-treatment: Polish the bare GCE with alumina slurry (e.g., 0.05 µm) on a microcloth, followed by sequential sonication in ethanol and deionized water to create a clean, reproducible surface.
- Electropolymerization: Immerse the cleaned GCE in an acidic solution containing ANSA monomer. Using cyclic voltammetry (CV), cycle the potential (e.g., between -0.5 V and +1.5 V vs. Ag/AgCl) for multiple scans (e.g., 10-15 cycles) to form a stable poly(ANSA) film on the GCE surface.
- Sensor Conditioning: Rinse the modified electrode (poly(ANSA)/GCE) with deionized water and cycle it in a clean supporting electrolyte (e.g., PBS) via CV until a stable baseline voltammogram is obtained.
- Dopamine Detection: Perform CV or Differential Pulse Voltammetry (DPV) in PBS containing various concentrations of dopamine. Record the oxidation peak current.
- Troubleshooting Tip: If the sensor response is unstable or low, ensure the electropolymerization solution is deoxygenated with an inert gas (e.g., N₂) and verify the number of polymerization cycles has been optimized.
- Objective: To synthesize an eco-friendly, non-enzymatic dopamine sensing material via a hydrothermal method.
- Materials: Nickel oxide (NiO) precursor, Zinc oxide (ZnO) precursor, date fruit extract.
- Step-by-Step Procedure:
- Solution Preparation: Dissolve precursors for NiO and ZnO in an appropriate solvent.
- Mixing and Capping: Add a specific volume of date fruit extract (e.g., 10, 15, 20 mL) to the mixture. The extract acts as a natural capping, stabilizing, and reducing agent, controlling the morphology of the resulting hybrid material.
- Hydrothermal Reaction: Transfer the solution to a Teflon-lined autoclave and heat it to a specific temperature (e.g., 120-180°C) for several hours.
- Product Isolation: After the reaction, allow the autoclave to cool naturally. Collect the resulting precipitate by centrifugation, and wash it several times with ethanol and deionized water. Dry the final product in an oven.
- Electrode Modification: Prepare an ink by dispersing the NiO/ZnO hybrid material in a solvent (e.g., ethanol/water) with a binder (e.g., Nafion). Drop-cast a measured volume of this ink onto the surface of a pre-cleaned GCE and allow it to dry.
- Troubleshooting Tip: The volume of date fruit extract critically influences the material's optical band gap and electrocatalytic properties. Systematically vary the volume and characterize the resulting material using XRD and SEM to identify the optimal synthesis condition.
Performance Data Comparison of Sensing Materials
Quantitative comparison of material performance allows for informed method selection. The following table consolidates key metrics from recent studies.
Table 2: Comparative Performance of Electrode Materials for Dopamine Detection
| Electrode Material | Modification/Renewal Technique | Detection Limit (LOD) | Linear Range | Selectivity Notes | Stability / Reproducibility |
|---|---|---|---|---|---|
| ECZO-MCPE [29] | Incorporation into carbon paste matrix | 0.455 µM | Not specified | Stable in sensing dopamine | 92% activity retained after 20 cycles |
| Poly(ANSA)/GCE [30] | Electropolymerization | 0.089 µM | 0.5 – 100 µM | Minimal interference from common analytes | Successfully applied to pharmaceutical samples |
| NiO/ZnO/GCE [31] | Drop-casting of hydrothermally synthesized hybrid | 0.036 µM | 0.01 – 4 mM | High selectivity demonstrated | Excellent repeatability and reproducibility |
| PEDOT-PPy/GCE [28] | Electropolymerized composite film | 5 nM | 5 nM – 200 µM | High selectivity in presence of interferents | Excellent reproducibility and stability |
| General MWCNTs [32] | Casting or growing on GCE surface | Varies with composite | Varies | Improved performance in composites | High stability; performance depends on functionalization |
Troubleshooting FAQs: Enhancing Reproducibility
Q1: Our dopamine oxidation peak current decreases significantly after a few measurement cycles. What is the most likely cause and how can it be mitigated?
- A: This is a classic sign of electrode fouling, caused by the adsorption of dopamine oxidation products (e.g., dopaminoquinone, which can form passivating polymeric films) onto the active electrode surface [27]. To mitigate this:
- Material Selection: Use materials known for anti-fouling properties. Conducting polymers like PEDOT-PPy [28] or composite materials like NiO/ZnO [31] have demonstrated excellent stability.
- Surface Renewal: Implement a consistent electrochemical renewal protocol between measurements. This often involves a series of CV scans in a clean supporting electrolyte to electrochemically clean the surface.
- Potential Window: Optimize the detection technique (e.g., using DPV instead of CV) to minimize the time the electrode is held at high oxidizing potentials.
Q2: How can I improve the selectivity of my sensor against common interferents like ascorbic acid (AA) and uric acid (UA)?
- A: Selectivity is achieved by creating an electrode interface that favors dopamine interaction. Key strategies include:
- Surface Charge: Use modification layers that are negatively charged (e.g., Nafion in PEDOT:Nafion [28] or the sulfonic groups in poly(ANSA) [30]) at physiological pH. This repels the anionic interferents (AA⁻ and UA⁻) while attracting the cationic dopamine.
- Pore Size/Size Exclusion: Nanostructured materials with tailored pore sizes can selectively allow dopamine molecules to access active sites while excluding larger or differently shaped interferents.
- Catalytic Specificity: Metal oxide hybrids (e.g., NiO/ZnO) provide electrocatalytic sites that preferentially lower the overpotential for dopamine oxidation compared to AA and UA [31].
Q3: What are the critical factors to control when renewing a solid electrode surface to ensure day-to-day reproducibility?
- A: Reproducibility hinges on strict adherence to a standardized renewal protocol. The key factors are:
- Mechanical Polishing: For bare GCEs, use a consistent polishing routine with defined alumina particle size, pressure, and pattern. Always follow with thorough sonication.
- Electrochemical Pre-treatment: For modified and bare electrodes, use a standardized electrochemical activation step (e.g., a set number of CV cycles in a specific electrolyte) to define the initial surface state.
- Modification Consistency: For drop-cast materials, control the concentration of the ink, the volume deposited, and the drying conditions (temperature, time, atmosphere).
- Documentation: Meticulously record the history of the electrode, including the number of uses and any renewal procedures applied. Establish a maximum usage limit for each electrode.
Logical Workflow and Signaling Pathways
The following diagram illustrates the logical decision-making process for selecting an appropriate electrode material and renewal technique based on experimental goals.
Decision Workflow for Sensor Design and Renewal
The fundamental signaling pathway involved in dopamine detection is its redox reaction. The electrochemical oxidation of dopamine is a 2-electron, 2-proton process, producing dopamine-o-quinone. This reaction is reversible, and the resulting reduction current can also be measured. The following diagram summarizes this core reaction pathway.
Dopamine Electrochemical Redox Reaction
Frequently Asked Questions (FAQs)
Q1: Why is electrode surface renewal critical for experimental reproducibility in electroanalysis? Electrode surfaces can become contaminated, passivated, or fouled during experiments, which severely undermines their analytical performance by reducing sensitivity and increasing background noise. Surface renewal processes restore the electrochemically active surface, ensuring consistent and reproducible results across experiments by re-establishing a well-defined electrode-electrolyte interface [33] [9].
Q2: What are the main methods for renewing solid electrode surfaces? The two primary methods are electrochemical and mechanical renewal:
- Electrochemical Renewal: Applies a specific potential or current in a suitable solution to clean the surface or introduce specific functional groups. This can be done in deionized water or electrolyte solutions [33].
- Mechanical Renewal: Involves physically refreshing the electrode surface in situ (without breaking the polarization circuit) to remove contaminants and oxides, providing a fresh, well-defined surface for measurement [9].
Q3: How do I choose the optimal electrochemical renewal parameters? The optimal parameters depend on your electrode material and target analyte. For instance, a carbon fiber microelectrode (CFME) can be effectively regenerated by applying 1.75 V for 26.13 minutes in deionized water. This treatment introduces oxygen-containing functional groups and significantly increases the electrochemical response to dopamine. Always consult literature for your specific system and validate the method with standard solutions [33].
Q4: What are common issues after electrode renewal and how can they be addressed? Common issues and their solutions are detailed in the table below.
Troubleshooting Guide
| Problem | Possible Causes | Recommended Solutions |
|---|---|---|
| High Background Noise | Residual impurities on surface Unsuitable renewal parameters Contaminated solution | Ensure thorough rinsing with pure solvent post-renewal Optimize potential and time for your system Use high-purity electrolytes and solvents [33] [34] |
| Poor Reproducibility Between Renewals | Inconsistent mechanical renewal force/pressure Varying renewal time Surface composition changes over time (for alloys) | Standardize the mechanical renewal procedure Use automated or controlled-force renewal devices Allow consistent equilibration time after renewal, especially for alloys [9] |
| Low Signal Sensitivity | Incomplete surface renewal Incorrect applied potential Electrode passivation | Verify renewal efficacy with a standard solution Perform a hydrodynamic voltammogram to find optimal detection potential Ensure renewal potential is sufficient to remove contaminants [33] [34] |
| Drifting Baseline | Slow surface equilibration post-renewal Unstable reference electrode | Allow adequate time for the electrode/solution interface to stabilize after applying potential Check the health and stability of your reference electrode [34] |
The following table summarizes specific operational parameters for different renewal methods as found in the literature.
Table 1: Electrochemical Renewal Parameters for Carbon Fiber Microelectrodes [33]
| Parameter | Specification | Effect / Note |
|---|---|---|
| Applied Potential | +1.75 V | vs. a suitable reference electrode |
| Treatment Time | 26.13 min | Can be optimized for specific equipment |
| Medium | Deionized Water | No added electrolyte required |
| Target Analyte | Dopamine | Used to validate renewal effectiveness |
| Key Outcome | Introduction of oxygen-containing functional groups, regeneration of electroactive surface | |
| Post-Renewal Performance | LOD for DA: 3.1 × 10⁻⁸ mol/L; Linear range: 1.0 × 10⁻⁷ to 1.0 × 10⁻⁴ mol/L (R² = 0.9961) |
Table 2: Considerations for Mechanically Renewed Alloy Electrodes [9]
| Factor | Description | Impact on Experiment |
|---|---|---|
| Equilibration Time | Surface composition changes over time after renewal due to surface segregation. | Electrochemical characteristics (e.g., capacitance) are time-dependent immediately after renewal. |
| Alloy Composition | Eutectic Sn-Pb (1 at.% Pb) studied. | Pb, as the surface-active component, segregates to the surface, changing interface properties. |
| Solvent | Acetonitrile (AN) solutions of LiClO₄. | Surface segregation kinetics are slower in AN than in aqueous solutions. |
Experimental Protocols
This protocol describes a simple method to regenerate and activate carbon fiber microelectrodes using only deionized water.
- Objective: To restore the electrochemical performance of an inactivated or contaminated carbon fiber microelectrode (CFME).
- Materials:
- Electrochemical Cell: Standard three-electrode system (Working Electrode: CFME; Counter Electrode: e.g., Pt wire; Reference Electrode: e.g., Ag/AgCl).
- Solution: High-purity deionized water.
- Potentiostat: To control and apply the potential.
- Procedure:
- Place the CFME, counter electrode, and reference electrode in a cell containing deionized water.
- Using the potentiostat, apply a constant potential of +1.75 V versus the reference electrode to the CFME.
- Maintain this potential for 26.13 minutes.
- After treatment, remove the electrode from the deionized water and rinse it. It can then be transferred to the analyte solution for testing.
- Validation: The success of regeneration can be validated by measuring the differential pulse voltammetry (DPV) response in a standard dopamine solution. A significantly increased current and a low detection limit confirm successful regeneration.
This protocol outlines how to investigate the surface segregation dynamics on a mechanically renewed solid alloy electrode.
- Objective: To monitor the evolution of the electrical double layer (EDL) structure on a renewed alloy surface over time.
- Materials:
- Electrode: Solid alloy electrode (e.g., Sn-Pb) with a mechanism for in-situ mechanical renewal.
- Electrochemical Cell: Three-electrode setup.
- Solution: Acetonitrile (AN) with LiClO₄ as a surface-inactive electrolyte.
- Impedance Analyzer or Potentiostat: For capacitance measurements.
- Procedure:
- Set up the electrochemical cell with the alloy electrode, counter electrode, and reference electrode in the AN solution.
- Perform in-situ mechanical renewal of the alloy electrode surface to create a fresh, well-defined interface.
- Immediately after renewal, begin a series of capacitance measurements (using impedance or cyclic voltammetry) at the determined zero-charge potential as a function of time.
- Continue these measurements over a period of minutes to hours to track the changes in the capacitance-potential curves.
- Data Analysis: The time-dependent changes in the capacitance curves indicate the surface segregation of the more surface-active component (e.g., Pb in a Sn-Pb alloy). The data can be fitted with kinetic models to understand the mechanism and rate of surface diffusion.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions and Materials
| Item | Function / Application |
|---|---|
| Deionized Water | Solvent for electrochemical renewal; effective without added electrolytes [33]. |
| Acetonitrile (AN) | Aprotic solvent for studies in non-aqueous systems; provides a wide potential window without solvent decomposition [9]. |
| Lithium Perchlorate (LiClO₄) | Surface-inactive electrolyte used in non-aqueous solutions for electrical double layer studies [9]. |
| Solid Alloy Electrodes (e.g., Sn-Pb) | Used to study surface segregation phenomena and time-dependent interface changes after renewal [9]. |
Experimental Workflow and Decision Pathway
The diagram below outlines a logical workflow for diagnosing electrode issues and selecting the appropriate surface renewal method to achieve consistent results.
Troubleshooting Renewal Processes and Optimizing for Long-Term Stability
Troubleshooting Guide
Why is my electrode regeneration incomplete, and how can I fix it?
Incomplete regeneration occurs when the electrode surface is not fully returned to its original, active state, often due to insufficient material removal or re-contamination.
- Diagnosis: Look for a drifting baseline, reduced peak resolution in cyclic voltammetry, or poor reproducibility in standard redox probes like ferricyanide.
- Solutions:
- Increase Removal Thickness: For mechanical cutter systems, ensure a sufficient layer (e.g., 4–5 μm for metallic electrodes and 0.1–1.5 μm for graphite-based electrodes) is removed to expose a fresh surface [7].
- Verify Cutter Performance: A worn-out cutter can cause inconsistent surface renewal. The cutter should be serviced or replaced after its serviceability limit (e.g., over 10^5 cuts) [7].
- Renew In-Situ: Perform the mechanical regeneration directly in the test solution to prevent air exposure from causing immediate surface passivation [7].
- Check for Leakage (Microfluidics): In PDMS-based microfluidic devices, ensure the elastomeric channels seal properly around the microwires during and after the renewal process to avoid sample leakage and contamination [4].
Why is my electrochemical response inconsistent after surface renewal?
Inconsistent responses typically stem from variations in the renewed surface's area, morphology, or composition.
- Diagnosis: High relative standard deviation (RSD) in repeated measurements of a standard analyte, or fluctuating background current.
- Solutions:
- Standardize Renewal Protocol: Ensure the renewal process (e.g., pulling microwires or activating the cutter) is performed in a highly reproducible manner, using automated systems where possible [4] [7].
- Calibrate Surface Area: After renewal, consistently verify the electroactive surface area using a standard redox couple.
- Inspect Microwires (Microfluidics): Manually sliding microwires can introduce variability. Check for bending, coating damage, or inconsistent sliding force [4].
- Validate Electrode Assembly: Confirm that all electrodes (working, counter, reference) are renewed simultaneously and that the pseudoreference electrode potential remains stable [4].
The table below consolidates key parameters from the literature for effective mechanical surface renewal.
| Parameter | Target Value / Specification | Applicable Electrode Types | Key Performance Metric |
|---|---|---|---|
| Layer Removal Thickness [7] | 4–5 μm | Pt, Ag, Au, Cu, Cd, Zn, Co, Ni (Metallic) | Fresh, electrochemically active surface |
| 0.1–1.5 μm | Graphite, Graphite-based | Fresh, electrochemically active surface | |
| Reproducibility [7] | RSD of 0.2–0.6% | All solid electrodes | Analytical signal over 10^4 renewal cycles |
| Cutter Service Life [7] | > 100,000 cuts | All solid electrodes | Consistent surface area and morphology |
| Flow Rate Tolerance [4] | Up to 40.0 mL min⁻¹ | Microfluidic microwire electrodes | Leak-free operation during/after renewal |
Experimental Protocol: Mechanical Renewal of Solid Electrodes
This detailed methodology is adapted from procedures for renewing electrode surfaces in situ using a mechanical cutter [7].
Apparatus Setup
- Electrode Assembly: Mount the solid indicator electrode (e.g., Pt, Ag, graphite) into the renewal device. Ensure the cutter tool (e.g., hexanite-R) is positioned at the correct angle for a clean cut.
- Solution Cell: Place the entire assembly into the electrochemical cell containing the test solution, ensuring the electrode surface is fully immersed.
Surface Renewal and Measurement
- Activation: Trigger the mechanical cutter to shave off a predefined surface layer (e.g., 4–5 μm for metals, 0.1–1.5 μm for graphite) [7].
- Immediate Testing: Without removing the electrode from the solution, initiate the electrochemical measurement (e.g., voltammetry). This "renewal in solution" prevents air-induced passivation and is critical for achieving high electrochemical activity [7].
The Scientist's Toolkit: Essential Research Reagents & Materials
| Item | Function |
|---|---|
| Polydimethylsiloxane (PDMS) | An elastomeric polymer used to fabricate microfluidic channels that form a tight seal around sliding microwires, enabling leak-free electrode renewal [4]. |
| Metal-Coated Microwires | Serve as renewable working, counter, and pseudoreference electrodes in microfluidic setups. They are slid through the channels to present a fresh surface [4]. |
| Mechanical Cutter (e.g., Hexanite-R) | A device used to precisely remove a thin surface layer (μm-scale) from a solid electrode in situ, restoring its electrochemical activity without chemical treatment [7]. |
| Standard Redox Probes (e.g., Ferri/Ferrocyanide) | Used to validate the success of the renewal process by checking for well-defined, stable voltammetric peaks and calculating the electroactive surface area. |
Workflow for Diagnosing Electrode Renewal Failures
The following diagram outlines a logical, step-by-step process for troubleshooting common issues with electrode renewal.
Frequently Asked Questions (FAQs)
What are the main advantages of renewable solid electrodes over traditional cleaning methods?
Renewable electrodes eliminate the need for chemical, physical, or mechanical surface treatments between measurements, which can add operational time, complexity, and introduce contaminants. The renewal process is fast, reproducible, and provides a fresh, consistent surface for each measurement, directly enhancing analytical precision and robustness [4].
Can this renewal method be used for high-voltage electroaccumulation?
Yes. The mechanical regeneration of the electrode surface directly in the solution is a primary requirement for depositing metals under high-voltage conditions (up to 1000 V) for electroaccumulation, as it ensures a clean and active surface at the start of the process [7].
My microwire electrodes in a PDMS device are leaking after renewal. What should I check?
The PDMS chip design is critical. A single-piece PDMS chip with interconnected channels relies on its elastomeric properties to seal around the microwires. Ensure the PDMS is properly cured and that the microwires are of the correct diameter. The design should allow pulling the microwires without leakage, even at flow rates up to 40.0 mL min⁻¹ [4].
How does in-situ renewal improve reproducibility in research?
In-situ renewal (renewing the surface while the electrode is immersed in the test solution) prevents the freshly exposed surface from being passivated by air before measurement. This is a key factor in enhancing the electrochemical activity and obtaining highly reproducible results across a vast number of measurement cycles (e.g., RSD of 0.2-0.6% over 10^4 cycles) [7].
Strategies to Mitigate Irreversible Deactivation and Catalyst Leaching
Troubleshooting Guides
Guide 1: Diagnosing and Addressing Catalyst Leaching in Liquid-Phase Reactions
Problem: A gradual, continuous decline in catalytic activity is observed in a liquid-phase batch reaction, along with detectable levels of catalytic metal in the product stream.
Explanation: This pattern suggests catalyst leaching, where active species detach from the solid support and enter the solution, causing irreversible activity loss and product contamination [35].
Troubleshooting Steps:
- Confirm Leaching: Analyze the reaction solution post-reaction using Inductively Coupled Plasma (ICP) spectroscopy to detect and quantify dissolved metal ions from the catalyst.
- Identify Root Cause:
- Weak Metal-Support Interaction: The bond between the active metal and the catalyst support is unstable under reaction conditions.
- Chemical Instability: The solid support (e.g., certain oxides or carbon) or the active phase itself dissolves in the reaction medium.
- Oxidative/Reductive Conditions: Reaction conditions lead to the formation of soluble metal complexes or ions.
- Implement Corrective Actions:
- Strengthen Metal-Support Interaction: Utilize catalysts designed with strong metal-support interaction (SMSI). This can be achieved by choosing appropriate support materials (e.g., certain forms of titania) or through catalyst pre-treatment protocols that enhance bonding [35].
- Modify Reaction Medium: Adjust the pH or solvent to conditions where the support and active metal are less soluble.
- Process Design: For continuous systems, consider implementing a downstream catalyst bed to capture leached species and protect the main catalyst charge.
Guide 2: Managing Irreversible Catalyst Poisoning
Problem: A sudden and permanent drop in catalyst activity occurs, and regeneration attempts (e.g., calcination) fail to restore performance.
Explanation: This is characteristic of irreversible poisoning, where contaminants form strong, stable chemical bonds with active sites, permanently blocking them [35] [36].
Troubleshooting Steps:
- Identify the Poison: Perform surface analysis on the spent catalyst (e.g., X-ray Photoelectron Spectroscopy - XPS) to detect specific elements like sulfur, potassium, or heavy metals on its surface [37] [35].
- Trace the Source: Check the feedstock for contaminant levels. In biomass conversion, for example, potassium or other metals can deposit on the catalyst and poison Lewis acid sites [37].
- Implement Corrective Actions:
- Feedstock Purification: Implement pre-treatment steps to remove poisons from the reactant stream before it contacts the catalyst [36].
- Select Poison-Tolerant Catalysts: Switch to a catalyst formulation whose active sites have a lower affinity for the identified poison. In some cases, adding catalyst promoters can confer resistance.
- Design for Regeneration: If poisoning is reversible, as with potassium on Pt/TiO₂ which can be removed via water washing, incorporate regular regeneration cycles into the process [37].
Guide 3: Overcoming Catalyst Fouling by Coke Deposition
Problem: Catalyst activity declines progressively over several reaction cycles. Regeneration by controlled combustion in air or oxygen restores most of the initial activity.
Explanation: This indicates fouling by coke—carbonaceous deposits that physically block active sites and pores [37] [35].
Troubleshooting Steps:
- Confirm Coke Formation: Use Thermogravimetric Analysis (TGA) on the spent catalyst. A significant weight loss upon heating in air between 300-500°C is indicative of coke combustion.
- Analyze Contributing Factors:
- Acidity: Strong acid sites on the catalyst promote coking.
- Diffusion Limitations: Low porosity or small pore diameters can trap coke precursors.
- Reaction Conditions: High temperature and low hydrogen partial pressure favor coke formation.
- Implement Corrective Actions:
- Optimize Catalyst Properties: Reduce excessive strong acid sites or use a catalyst with a hierarchical pore structure to improve mass transfer and reduce pore blockage [35].
- Employ the Metal-H₂ Method: For solid acid catalysts, modifying the catalyst with a transition metal (e.g., Pt, Co) and operating under a H₂ atmosphere can gasify coke precursors as they form, significantly improving stability [35].
- Optimize Process Parameters: Increase hydrogen partial pressure or lower the reaction temperature to thermodynamically disfavor coke formation [35].
Frequently Asked Questions (FAQs)
Q1: What is the fundamental difference between reversible and irreversible catalyst deactivation?
A1: Reversible deactivation, such as certain types of coking or fouling, can be counteracted through in-situ or ex-situ regeneration protocols (e.g., calcination in air, hydrogen treatment) to restore most of the catalyst's original activity [35]. Irreversible deactivation, which includes mechanisms like strong poisoning, leaching, or sintering, causes permanent damage that cannot be economically repaired, necessitating catalyst replacement [35] [36].
Q2: How can I experimentally determine if my catalyst is sintering?
A2: Sintering, the growth of active metal particles leading to reduced surface area, is best confirmed through characterization techniques that probe catalyst morphology. Transmission Electron Microscopy (TEM) provides direct visual evidence of metal particle size and distribution. Chemisorption (e.g., H₂ or CO chemisorption) quantitatively measures the dispersion and active surface area of the metal, with a decrease indicating sintering [36].
Q3: Are there strategies to prevent catalyst deactivation at the research and design stage?
A3: Yes, proactive strategies are highly effective. Consider deactivation mechanisms early in catalyst design [37]. Key approaches include:
- Strengthening Metal-Support Interaction: Select supports and pre-treatment methods that create strong bonds to anchor metal particles and resist sintering [35].
- Designing Pore Architecture: Use supports with optimized pore networks (e.g., hierarchical pores) to minimize diffusion limitations and reduce fouling [35].
- Conducting Extended-Duration Tests: Move beyond short-term "break-in" period tests to perform long-term stability experiments that reveal slow deactivation processes [37].
Q4: What is the "Metal-H₂ method" for controlling deactivation?
A4: This is an effective strategy for stabilizing solid acid catalysts (e.g., in dehydration, condensation reactions). It involves modifying the solid acid with a transition metal (e.g., Pt, Co) and carrying out the reaction under a hydrogen atmosphere. The metal facilitates the activation of H₂, which helps to hydrogenate and remove carbonaceous deposits (coke precursors) from the acid sites as they form, thereby maintaining catalyst activity over extended periods [35].
Table 1: Common Catalyst Deactivation Mechanisms and Mitigation Strategies
| Mechanism | Primary Cause | Key Mitigation Strategy | Recoversbility |
|---|---|---|---|
| Sintering [35] [36] | High temperature causing particle growth | Use supports with Strong Metal-Support Interaction (SMSI); lower operating temperature | Often Irreversible |
| Poisoning [37] [35] [36] | Strong chemisorption of contaminants (e.g., S, K) | Purify feedstock; design poison-tolerant catalysts | Reversible or Irreversible depending on poison strength |
| Fouling/Coking [37] [35] | Blockage by carbonaceous deposits | Use Metal-H₂ method; optimize pore structure; periodic regeneration with air/oxygen | Typically Reversible |
| Leaching [35] | Detachment of active species into solution | Strengthen metal-support bond; modify reaction medium (pH/solvent) | Irreversible |
Table 2: Research Reagent Solutions for Catalyst Deactivation Studies
| Reagent / Material | Function in Experiment | Application Context |
|---|---|---|
| Pt/TiO₂ Catalyst [37] | Model catalyst for studying poisoning mechanisms. Used to investigate the effect of contaminants like potassium on Lewis acid sites. | Biomass conversion, catalytic fast pyrolysis |
| Indium Foil & Li Metal [15] | Components for forming an alloy negative electrode in all-solid-state battery testing, relevant for interfacial stability studies. | Solid-state battery research, electrode interface studies |
| Li₆PS₅Cl Solid Electrolyte [15] | A sulfide-based solid electrolyte used to study interfacial reactions and degradation at the electrode-electrolyte boundary. | Solid-state battery research, ionic conductivity studies |
| Polydimethylsiloxane (PDMS) Microfluidic Chip [4] | Platform for implementing renewable solid electrodes by sliding metal-coated microwires, eliminating need for electrode cleaning. | Electroanalytical chemistry, surface renewal studies |
Experimental Protocols
Protocol 1: Testing Catalyst Stability Against Leaching
Objective: To quantitatively assess the resistance of a solid catalyst to leaching of its active components in a liquid-phase reaction.
Materials:
- Catalyst sample
- Reactant solution
- Batch reactor (e.g., glass flask with condenser)
- Heating/stirring mantle
- Syringe filter (0.2 µm)
- ICP-OES/MS instrument
Methodology:
- Reaction Setup: Charge the reactor with a known mass of catalyst and reactant solution under inert atmosphere.
- Run Reaction: Conduct the reaction at predetermined conditions (temperature, time) with constant agitation.
- Sample Separation: After the reaction, cool the mixture and rapidly separate the solid catalyst from the liquid product using centrifugation and syringe filtration.
- Analysis: Acidify an aliquot of the clear filtrate and analyze it using ICP-OES or ICP-MS to quantify the concentration of leached metal ions.
- Correlation: Measure the catalytic conversion/activity and correlate it with the amount of leached metal to determine the stability of the catalyst.
Protocol 2: Accelerated Aging Test for Coke Formation
Objective: To simulate and study long-term coke formation on a catalyst under controlled, accelerated conditions.
Materials:
- Catalyst sample
- Tubular quartz reactor
- Furnace with temperature control
- Mass flow controllers
- Feedstock gas/liquid (e.g., hydrocarbon)
- Thermogravimetric Analyzer (TGA)
Methodology:
- Catalyst Loading: Place a known mass of fresh catalyst in the reactor.
- Accelerated Aging: Expose the catalyst to a concentrated stream of coke-forming feedstock (e.g., propylene or 1,3-butadiene) in an inert carrier gas at an elevated temperature for a fixed duration.
- Quantification of Coke: Unload the coked catalyst and analyze it using TGA. Heat the sample in air to combust the carbon deposits; the weight loss corresponds to the amount of coke formed.
- Activity Assessment: Test the catalytic activity of the coked sample and compare it to the fresh catalyst to quantify the deactivation.
Workflow and Pathway Diagrams
Catalyst Deactivation Diagnosis
Metal-H₂ Coke Removal Mechanism
Energy Efficiency Considerations in Mechanical and Electrochemical Renewal Systems
Troubleshooting Guide: Frequently Asked Questions
1. What are the most common signs that my solid electrode surface needs renewal?
A decrease in electrochemical sensitivity or resolution, along with poor reproducibility between measurements, are the primary indicators that an electrode surface has become contaminated, passivated, or fouled. This manifests as a drift in the baseline signal or a diminishing voltammetric peak current for your standard analyte [38] [33]. For example, in renewable solid electrodes using microwires, the need for renewal arises when the surface morphology or composition is nonspecifically changed, undermining analytical performance [4].
2. I am prioritizing energy efficiency in my lab. Which electrode renewal method should I choose?
For the highest energy efficiency, mechanical renewal methods are generally superior. Techniques like sliding metal-coated microwires or in-situ auto-renewal devices consume minimal electrical energy as they primarily rely on mechanical motion to expose a fresh surface [4] [38]. In contrast, electrochemical regeneration often requires applying a specific potential for a prolonged period (e.g., 1.75 V for 26 minutes), which consumes more direct electrical energy [33]. However, the optimal choice also depends on your application constraints, such as the need for a closed system or compatibility with microfluidics.
3. How can I minimize energy consumption during the electrochemical regeneration of carbon fiber microelectrodes?
A key strategy is to use deionized water as your activation medium instead of solutions containing electrolytes. Research has demonstrated that effective regeneration can be achieved in deionized water, which reduces the need for chemical preparation and purification, indirectly contributing to lower overall energy consumption in lab operations [33]. Furthermore, optimizing the activation parameters—potential application time (26.13 min in one cited case) and potential value (1.75 V)—to the minimum required for satisfactory performance will directly reduce energy usage [33].
4. My post-renewal signals are inconsistent. How can I improve reproducibility?
Poor reproducibility often stems from inconsistent renewal protocols. To address this:
- Standardize the Protocol: For mechanical renewal, ensure the sliding distance, speed, and pressure on the microwires are consistent [4]. For electrochemical renewal, strictly control the applied potential and duration [33].
- Validate the Surface: Use a standard redox probe like Fe(II)/Fe(III) to verify that the renewed electrode produces a consistent peak current and potential before running your actual samples [38].
- Check Material Integrity: Ensure that the renewal process itself (e.g., repeated pulling of microwires or electrochemical treatment) is not degrading the electrode material over time.
5. What are the energy trade-offs between frequent in-situ renewal and batch-mode polishing?
Frequent in-situ renewal, as part of an automated analytical workflow, can be more energy-efficient on a per-measurement basis. It eliminates the need for separate, often manual, polishing steps that require additional equipment and operator time, thereby reducing the overall energy footprint of the analysis process [4] [38]. Batch polishing of multiple electrodes offline may seem less energy-intensive for the renewal act itself, but it incurs higher labor and overhead costs, making the overall process less efficient.
Troubleshooting Common Problems
The table below summarizes specific issues, their probable causes, and energy-conscious solutions.
| Problem | Probable Cause | Energy-Efficient Solution |
|---|---|---|
| Low Signal Intensity | Contaminated or passivated electrode surface; Inefficient renewal. | Employ in-situ mechanical renewal (e.g., slide microwires) to restore activity without chemicals or external energy for polishing [4]. |
| Poor Reproducibility | Inconsistent renewal between experiments; Variable surface area. | Adopt an automated in-situ renewal device to ensure identical surface exposure each time, reducing energy wasted on repeated experiments [38]. |
| Baseline Drift | Slow surface fouling during measurement; Unstable regeneration. | Implement a pre-emptive, scheduled renewal cycle integrated into your microfluidic method to maintain a stable baseline [4] [39]. |
| High Electrical Consumption | Use of energy-intensive electrochemical regeneration protocols. | Optimize electrochemical parameters (potential, time) or switch to low-energy mechanical renewal where applicable [38] [33]. |
Standard Experimental Protocols
Protocol 1: Mechanical Renewal of Solid Electrodes in Microfluidics
This protocol details the procedure for renewing electrode surfaces by sliding metal-coated microwires, a highly energy-efficient method suitable for integrated microfluidic systems [4].
1. Principle The electrochemical activity of a solid electrode is recovered by manually sliding a metal-coated microwire through a polydimethylsiloxane (PDMS) microchannel. This action exposes a fresh, uncontaminated electrode surface without any chemical treatment or polishing, analogous to the function of a mercury drop electrode [4].
2. Required Materials and Reagents
- PDMS Microfluidic Chip: A single-piece chip with parallel channels for electrodes and a perpendicular top channel.
- Metal-Coated Microwires: Function as working, counter, and pseudoreference electrodes.
- Sample Solution: The solution to be analyzed.
3. Step-by-Step Procedure 1. Assembly: Insert the metal-coated microwires into the parallel channels of the PDMS chip, designating them as working, counter, and pseudoreference electrodes. 2. Introduction of Sample: Flow the sample solution through the top, perpendicular channel. 3. Electrochemical Measurement: Perform the desired voltammetric measurement (e.g., cyclic voltammetry). 4. Surface Renewal: To recover electrochemical activity, simply pull the microwires manually to slide a fresh segment into the measurement zone. The elastomeric nature of PDMS prevents leakage during this process, even at flow rates up to 40.0 mL/min [4]. 5. Repeat: Conduct subsequent measurements on the renewed surface.
Protocol 2: Electrochemical Regeneration of Carbon Fiber Microelectrodes in Deionized Water
This protocol describes a method to regenerate and activate carbon fiber microelectrodes using an electrochemical treatment in deionized water, restoring sensitivity for analytes like dopamine [33].
1. Principle An applied anodic potential in deionized water induces electrochemical surface modifications on the carbon fiber, regenerating the electrochemically active surface. This is attributed to the introduction of oxygen-containing functional groups that enhance electron transfer kinetics [33].
2. Required Materials and Reagents
- Carbon Fiber Microelectrode (CFME): The electrode to be regenerated.
- Deionized Water: The activation medium. No additional electrolytes are needed.
- Potentiostat: Equipment to apply a controlled potential.
- Standard Dopamine Solutions: For validating the regeneration efficacy.
3. Step-by-Step Procedure 1. Setup: Immerse the inactivated or contaminated CFME in deionized water. 2. Electrochemical Treatment: Apply an anodic potential of +1.75 V for a duration of 26.13 minutes [33]. 3. Rinsing: Gently rinse the regenerated CFME with clean deionized water. 4. Validation: Test the electrochemical performance of the regenerated CFME using a standard such as dopamine. A differential pulse voltammetry (DPV) response with good linearity (R² = 0.9961) from 1.0 × 10⁻⁷ to 1.0 × 10⁻⁴ mol/L dopamine confirms successful regeneration [33].
Experimental Workflow and Decision Pathway
The diagram below outlines a logical workflow for selecting and implementing an energy-efficient electrode renewal strategy based on experimental requirements.
Research Reagent and Material Solutions
The following table lists key materials and reagents essential for implementing the described electrode renewal methods.
| Item | Function/Application | Energy Efficiency Consideration |
|---|---|---|
| Polydimethylsiloxane (PDMS) Chip | Elastomeric microfluidic device that enables leak-free sliding of microwires for in-situ mechanical renewal [4]. | Eliminates energy consumption for electrode cleaning and polishing, central to low-energy, continuous sensing platforms. |
| Metal-Coated Microwires | Serve as renewable working, counter, and reference electrodes within microfluidic channels [4]. | Their renewal is a manual or low-power process, avoiding energy-intensive electrochemical pre-treatments. |
| Deionized Water | A chemical-free medium for the electrochemical regeneration of carbon fiber microelectrodes [33]. | Reduces energy footprint associated with production, disposal, and purification of chemical electrolytes. |
| Alumina or Diamond Polish | Abrasive material for traditional mechanical polishing of glassy carbon and other solid electrodes [38]. | The polishing process itself is low energy, but it often requires subsequent cleaning steps, increasing the total energy and time cost. |
| Standard Fe(II) Solutions | Used with 2,2'-bipyridyl to validate the surface condition and reproducibility of a renewed electrode [38]. | Ensures analytical performance, preventing energy waste on experiments conducted with poorly performing electrodes. |
Frequently Asked Questions (FAQs)
Q1: What are the primary environmental factors that accelerate biofouling on submerged sensors? Biofouling initiation and rates are heavily influenced by environmental conditions. Key factors include temperature, salinity, nutrient availability, and hydrodynamics [40]. Tropical zones, with higher biological growth rates, experience more intense and rapid biofouling compared to colder regions [40]. Furthermore, surface properties like roughness and electrostatic charge significantly impact the initial adhesion of organisms [40].
Q2: Why does protein adsorption often lead to a loss of protein bioactivity? Adsorption-induced loss of bioactivity can result from two main factors:
- Conformational Changes: The interaction with the surface can cause the protein to unfold, distorting the geometry of its bioactive site [41].
- Steric Hindrance: The protein may adsorb in an orientation that physically blocks access to its bioactive site, preventing ligands from binding [41]. Standard bioactivity assays cannot distinguish between these causes, requiring more detailed structural analysis.
Q3: What are the most promising, environmentally sustainable strategies for controlling biofouling? Research indicates a shift towards integrated, eco-friendly solutions. The most promising strategies involve a combination of physical, chemical, and biological methods integrated with sustainable coatings [40]. This includes the development of degradable materials, natural antifoulants from marine organisms, and smart responsive coatings that react to environmental stimuli [42]. There is a strong trend and regulatory push to move away from traditional toxic biocides [40].
Q4: How can I determine if a loss in bioactivity is due to protein unfolding or orientation? A single technique is insufficient. A multi-technique approach is required to provide a complementary dataset [41] [43]:
- Use Adsorbed-State Circular Dichroism (CD) to quantify adsorption-induced changes in protein secondary structure [41].
- Use Amino-Acid Labeling/Mass Spectrometry (AAL/MS) to identify regions of the protein that are sterically blocked by the surface (indicating orientation) or have become unfolded and exposed [41].
- Correlate these findings with bioactivity assays to identify the root cause.
Troubleshooting Guides
Problem: Inconsistent Protein Adsorption Results
| Symptom | Possible Cause | Solution |
|---|---|---|
| Variable surface coverage between experiments. | Inconsistent surface preparation or contamination. | Implement a strict, documented surface cleaning and validation protocol before each experiment. |
| Unpredictable loss of protein function. | Adsorption-induced denaturation or non-optimal orientation. | Characterize the adsorbed protein layer using techniques like CD or AAL/MS to understand the structural changes [41]. Consider using a different surface chemistry or a tethering ligand to control orientation. |
| Irreversible protein binding complicating surface renewal. | High adsorption energy and strong surface-protein interactions. | Protein desorption is inherently slow; the Gibbs' adsorption energy for proteins is 2–6 orders of magnitude larger than for typical surfactants [44]. Focus on preventive strategies or use cleaning solutions designed to disrupt protein-surface bonds. |
Problem: Rapid Biofouling on Marine Sensor Surfaces
| Symptom | Possible Cause | Solution |
|---|---|---|
| Data accuracy degrades within days/weeks of deployment. | Formation of biofilm and attachment of macro-foulers (e.g., barnacles, algae) [42]. | Apply an eco-friendly antifouling coating. Research is active in coatings with natural antifoulants, fouling-release surfaces, and smart coatings that degrade [40] [42]. |
| Increased measurement error (e.g., for CTD sensors). | Biofouling on sensor probes (conductivity, optical windows) [42]. | Integrate a mechanical wiper or use a biofou-resistant material (e.g., copper alloy) for the sensor face, where possible without interfering with measurements. |
| Corrosion of metallic sensor housings. | Microbiologically Influenced Corrosion (MIC), often from Sulfate-Reducing Bacteria (SRB) in biofilms [42]. | Ensure cathodic protection systems are functional and consider coatings that resist SRB adhesion. |
Experimental Protocols
Protocol 1: Characterization of Adsorbed Protein Structure and Orientation
Objective: To determine the secondary structure and solvent accessibility of a protein adsorbed onto a solid surface.
Materials:
- Solid substrate of interest (e.g., fused silica, polymer film)
- Protein solution in appropriate buffer
- Adsorbed-state Circular Dichroism (CD) spectropolarimeter
- Reagents for Amino-Acid Labeling (AAL) (e.g., succinic anhydride, NHS-acetate)
- Mass Spectrometry (MS) system
Methodology:
- Surface Preparation: Clean and characterize the substrate surface to ensure reproducibility.
- Protein Adsorption: Incubate the substrate in the protein solution for a defined period. Use a low solution concentration to allow for protein unfolding and spreading, or a high concentration to promote competitive adsorption and limited unfolding [41] [44].
- Circular Dichroism (CD):
- Place the protein-adsorbed substrate in the CD spectropolarimeter.
- Collect spectra in the far-UV range (e.g., 190-250 nm) to determine the percentages of α-helix, β-sheet, and random coil structures [41].
- Compare to the CD spectrum of the protein in solution to identify adsorption-induced structural changes.
- Amino-Acid Labeling/Mass Spectrometry (AAL/MS):
- Perform chemical labeling on the native protein in solution and separately on the adsorbed protein using a mild labeling reagent.
- Digest the labeled proteins into peptides.
- Analyze the peptide fragments using MS to identify the specific amino acid residues that were labeled.
- Interpretation: Residues labeled in solution but not after adsorption indicate regions blocked by the surface (orientation). Residues unlabeled in solution but labeled after adsorption indicate areas that have unfolded and become exposed [41].
Protocol 2: Evaluating Eco-Friendly Antifouling Coatings
Objective: To test the efficacy of a novel, non-toxic coating in preventing marine biofouling.
Materials:
- Coated and uncoated (control) test panels
- Marine testing site (e.g., dock, buoy system)
- Digital camera or microscope for documentation
- Image analysis software (e.g., ImageJ)
Methodology:
- Experimental Setup: Securely mount coated and control panels on a rack. Ensure they are submerged at a consistent depth and orientation, facing the primary water flow.
- Long-Term Deployment: Deploy panels for a defined period (e.g., 1, 3, 6, 12 months) to capture seasonal variations in fouling communities [40].
- Monitoring and Data Collection:
- Retrieve panels at regular intervals.
- Photograph panels using a standardized setup.
- Quantify fouling coverage by analyzing images to determine the percentage of surface area covered by biofouling.
- Identify and count key fouling organisms (e.g., barnacles, tubeworms, algae).
- Data Analysis: Compare the fouling coverage and organism density on the coated panels versus the control panels. Statistical analysis (e.g., t-test) should be used to confirm the significance of the coating's performance.
Data Presentation
Table 1: Comparison of Techniques for Characterizing Surface-Bound Proteins
| Technique | Information Provided | Key Advantage | Key Limitation |
|---|---|---|---|
| Quartz Crystal Microbalance with Dissipation (QCM-D) | Adsorption mass ("wet mass" including hydrodynamically coupled water), viscoelastic properties [43]. | Real-time, label-free kinetics. | Lacks chemical specificity; measures hydrated mass. |
| Surface Plasmon Resonance (SPR) | Adsorption mass ("dry mass" of biomolecule), kinetics, and binding affinities [43]. | Real-time, label-free kinetics; high sensitivity. | Lacks chemical specificity; signal is sensitive to temperature. |
| X-ray Photoelectron Spectroscopy (XPS) | Elemental and chemical state composition of the top ~10 nm; quantitative [43]. | Provides quantitative atomic concentration data. | Requires Ultra-High Vacuum (UHV); expert interpretation needed. |
| Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS) | Molecular structure and spatial distribution of surface species; high surface sensitivity (top 1-2 nm) [43]. | Extremely high surface sensitivity; rich molecular information. | Semi-quantitative; requires UHV; complex data analysis. |
| Circular Dichroism (CD) | Protein secondary structure (α-helix, β-sheet content) [41]. | Probes conformational changes directly. | Typically requires transparent, flat substrates. |
| Amino-Acid Labeling/Mass Spectrometry (AAL/MS) | Adsorbed protein orientation and tertiary structure unfolding [41]. | Provides residue-level structural information. | Destructive; complex sample preparation. |
Table 2: Impact of Biofouling on Different Marine Equipment
| Equipment Type | Key Impacts of Biofouling | Performance & Cost Consequences |
|---|---|---|
| Marine Vessels | Increased hull roughness and hydrodynamic drag [42]. | Fuel consumption increase of 9-84%; global extra fuel cost of ~$56M for naval fleets [42]. |
| Tidal/Wave Turbines | Reduced lift coefficient, increased drag on blades [42]. | 1mm of fouling can reduce lift coefficient by ~15% and lift-to-drag ratio by up to 90% [42]. |
| Offshore Platforms | Increased structural weight, accelerated corrosion, fatigue [42]. | 250mm fouling layer can reduce platform fatigue life by 54.0% [42]. |
| Sensors & ROVs | Signal distortion, mechanical jamming, optical signal attenuation [42]. | CTD sensor failure within 2 weeks; wave buoy data errors >30% [42]. |
Research Reagent Solutions
Table 3: Essential Materials for Biofouling and Protein Adsorption Research
| Item | Function/Application |
|---|---|
| Transparent Planar Substrates (e.g., Fused Silica, PMMA) | Essential for optical characterization techniques like Adsorbed-State CD to study protein structure on surfaces [41]. |
| Functionalized Biosensor Chips (e.g., SPR, QCM-D) | Gold or silica chips, often pre-coated with carboxyl or amine groups, for immobilizing biomolecules and studying adsorption kinetics in real-time [43]. |
| Eco-Friendly Antifouling Coating Formulations | Coatings incorporating natural biocides (e.g., from marine organisms), fouling-release polymers, or degradable materials to prevent fouling without toxicity [40] [42]. |
| Triply Periodic Minimal Surface (TPMS) Structures | 3D-printed porous electrodes with optimized geometry to enhance mass/fluid transport and provide active reaction sites, minimizing fouling-prone stagnation zones [45]. |
| Mild Labeling Reagents for AAL (e.g., Succinic Anhydride) | Chemicals that covalently modify specific amino acid side chains (e.g., lysine) under mild conditions to probe solvent accessibility in protein structure studies [41]. |
Workflow and Strategy Diagrams
Benchmarking Performance: Validating and Comparing Renewal Method Efficacy
Frequently Asked Questions (FAQs)
Q1: What are the key quantitative metrics I should report to demonstrate the performance of my electrochemical sensor? You should primarily report the Limit of Detection (LOD), sensitivity, and reproducibility (often as Relative Standard Deviation, RSD). The LOD defines the lowest analyte concentration your sensor can reliably detect, while sensitivity is the slope of the calibration curve. Reproducibility confirms the reliability of your measurements across multiple electrodes or renewals. [46] [47]
Q2: My sensor's signal is unstable. Could this be related to my surface renewal technique? Yes, inconsistent surface renewal is a major cause of signal drift. A poorly renewed surface can have variable uncompensated resistance and active surface area, leading to fluctuating signals. Ensure your renewal protocol—whether mechanical polishing, electrochemical activation, or a regenerative mechanism—is performed meticulously and consistently before each measurement. [46] [48] [49]
Q3: How can I improve the reproducibility of my solid electrode, especially after surface renewal? Utilize a fixed-geometry electrode design that integrates all electrodes into a single unit. This minimizes user-dependent positioning errors and variable uncompensated resistance, which are significant sources of irreproducibility in traditional dipping methods. [46] Furthermore, establish a strict, validated protocol for surface pre-treatment. [49]
Q4: What is an acceptable RSD value for a high-quality voltammetric sensor? For a portable device, an RSD of ≤ 0.90% is considered excellent analytical reproducibility. In studies of renewable electrodes, RSD values around 2.39% have been reported for low analyte concentrations, indicating good repeatability. [46] [47]
Q5: Why is my modified electrode's performance declining over multiple uses? Performance decay is often due to sensor fouling and irreversible adsorption of contaminants or reaction products. Implementing a regenerative mechanism that advances fresh sensing material or provides a consistent renewal process (like mechanical polishing) can restore the electrode's active surface and ensure high operational durability. [46] [48]
Troubleshooting Guides
Issue 1: High Background Noise and Unstable Baseline
| Possible Cause | Solution |
|---|---|
| Contaminated electrode surface from previous experiments. | Implement a rigorous cleaning protocol: mechanically polish with alumina slurry and then electrochemically clean in a supporting electrolyte by cycling over the intended potential window until a stable voltammogram is obtained. [48] [49] |
| Un-optimized or unstable reference electrode. | Use a stable reference electrode (e.g., Ag/AgCl) and ensure it is properly maintained. In advanced designs, an integrated, fixed-geometry reference electrode within a single cartridge can prevent these issues. [46] [50] |
Issue 2: Poor Reproducibility Between Measurements or Electrodes
| Possible Cause | Solution |
|---|---|
| Inconsistent inter-electrode positioning in traditional three-electrode setups. | Transition to an integrated electrode system where the working, reference, and counter electrodes are housed in a fixed-geometry cartridge. This eliminates positional variability as a source of error. [46] |
| Inconsistent surface renewal. For carbon paste electrodes, this could be uneven packing or polishing. | For renewable electrodes, use a controlled, automated mechanism. For example, a piston-driven system that advances fresh material or a mechanical polisher with fixed pressure and time can yield a regeneration efficiency of >99%. [46] [47] |
Issue 3: Lower-than-Expected Sensitivity and Higher LOD
| Possible Cause | Solution |
|---|---|
| Insufficient electroactive surface area or poor electron transfer kinetics. | Apply appropriate surface treatments to your electrode. Chemical treatments (e.g., in NaOH or HNO3) or electrochemical activation can expose more conductive material and introduce functional groups, enhancing sensitivity. [49] [51] |
| Suboptimal chemical modification of the electrode surface. | Re-optimize the modification process. For instance, when using a Molecularly Imprinted Polymer (MIP), ensure the correct functional monomer-to-template ratio is used to create effective recognition sites. [46] |
Quantitative Performance Data from Recent Studies
The following table summarizes key performance metrics from recent studies, providing benchmarks for sensor assessment.
Table 1: Quantitative Metrics from Electrochemical Sensor Studies
| Sensor Platform / Electrode Type | Analyte | Limit of Detection (LOD) | Reproducibility (RSD) | Key Innovation for Reproducibility |
|---|---|---|---|---|
| Pen-like trielectrode integrated system [46] | Salicylic Acid (ScA) | 1.06 µM | ≤ 0.90% | Fixed-geometry housing & regenerative piston mechanism |
| Renewable silver-based mercury film electrode (Hg(Ag)FE) [47] | Brilliant Blue FCF (BB) | 0.24 µg L⁻¹ | 2.39% (n=6) | Mechanical surface renewal before each measurement |
| Cerium-doped Fe₂O₃ modified carbon paste electrode (Ce–Fe₂O₃/CPE) [51] | Thymol (TML) | 14.05 nM | Information in source paper | Nanomaterial-enhanced sensitivity and stability |
Detailed Experimental Protocols
Protocol 1: Fabrication and Operation of a Regenerative Pen-like Electrode System
This protocol is adapted from the integrated trielectrode system designed for superior reproducibility. [46]
- Cartridge Assembly: Integrate the working, reference (Ag/AgCl), and counter (carbon) electrodes into a single, disposable cartridge. The spatial relationship between electrodes must be fixed and invariable.
- Working Electrode Loading: Load the sensing material (e.g., a Carbon Black-PLA composite or a MIP) into the cartridge's piston-driven refill mechanism.
- Surface Regeneration: Activate the regenerative piston to advance fresh sensing material to the electrode tip, discarding the used layer. This ensures a consistent electroactive surface for each measurement.
- Measurement: Immerse the pen-like device into the sample solution and perform voltammetric analysis (e.g., DPV, SWV).
- Validation: After a set number of measurements, validate sensor performance by checking the recovery percentage of a standard analyte solution. The system should achieve a Regeneration Efficiency (RE) of >99%.
Protocol 2: Surface Treatment of 3D-Printed Carbon-Based Electrodes
This protocol outlines chemical treatments to enhance the performance of lab-made electrodes. [49]
- Electrode Fabrication: Manufacture electrodes using a conductive filament (e.g., Carbon Black and Polylactic Acid composite) in a 3D printing pen or printer.
- Mechanical Polishing: Initially polish the electrode surface with 320-grit wet sandpaper to achieve a smooth and uniform baseline.
- Chemical Treatment (Basic Medium - Recommended):
- Immerse the electrode in a 1.00 mol L⁻¹ sodium hydroxide (NaOH) solution for 30 minutes.
- Remove the electrode, wash it thoroughly with ethanol and purified water, and allow it to dry at room temperature for 12 hours.
- Alternative Treatments:
- Acidic Treatment: Immerse in 7.90 mmol L⁻¹ nitric acid (HNO₃) for 15 minutes.
- Solvent Treatment: Immerse in dimethylformamide (DMF) for 15 minutes.
- Electrochemical Characterization: Characterize the treated electrode using Cyclic Voltammetry (CV) and Electrochemical Impedance Spectroscopy (EIS) in a solution containing a redox probe (e.g., 5.00 mmol L⁻¹ ferricyanide/ferrocyanide in 0.10 M KCl) to determine the improved electroactive area and electron transfer kinetics.
Research Reagent Solutions: Essential Materials
This table lists key materials used in the featured experiments to achieve high reproducibility.
Table 2: Essential Research Reagents and Materials
| Item | Function / Application | Example from Research |
|---|---|---|
| Ag/AgCl Reference Electrode | Provides a stable and reproducible reference potential for accurate voltammetric measurements. | Used in the pen-like integrated system and with the Hg(Ag)FE. [46] [47] |
| Carbon Black / Polylactic Acid (PLA) Composite | Serves as a low-cost, conductive material for fabricating lab-made and 3D-printed electrodes. | Used as the base material for the working electrode in 3D-printed sensors. [49] |
| Molecularly Imprinted Polymer (MIP) | Acts as a synthetic receptor on the electrode surface, providing high selectivity for a specific target analyte. | Used for the selective detection of Salicylic Acid (ScA) in the pen-like system. [46] |
| Cerium-doped Iron Oxide (Ce–Fe₂O₃) Nanoparticles | Used as a nanomaterial modifier to enhance the electrocatalytic properties and sensitivity of carbon paste electrodes. | Modifier for the voltammetric detection of thymol. [51] |
| Dimethylformamide (DMF) & Sodium Hydroxide (NaOH) | Chemical agents for the post-fabrication surface treatment of electrodes, helping to expose conductive sites and improve electron transfer. | Used to treat the surface of 3D-printed CB-PLA electrodes. [49] |
Workflow and Relationship Diagrams
Diagram 1: Experimental workflow for reproducible sensor development.
Diagram 2: Key strategies for achieving reproducibility.
Troubleshooting Guides & FAQs
Frequently Asked Questions
Q1: What is the core difference between electrochemical and mechanical surface renewal, and why does the choice matter for my research?
A1: The core difference lies in the mechanism of creating a fresh, reproducible electrode surface.
- Mechanical Renewal physically removes a thin contaminated surface layer (e.g., via polishing or cutting) to expose a pristine bulk material underneath. This is highly effective for removing adsorbed impurities and oxides [52] [53].
- Electrochemical Renewal uses applied potentials in a clean electrolyte to drive redox reactions that desorb contaminants or reduce surface oxides without physical abrasion [54].
The choice is critical for reproducibility. Mechanical renewal standardizes the initial surface state but may introduce abrasives or strain. Electrochemical renewal is non-contact but efficacy depends heavily on the electrode material, the nature of contamination, and the selected potential window. Selecting the wrong method can lead to inconsistent surface states and unreproducible data [53] [54].
Q2: My data shows high variability even after polishing. What could be going wrong with my mechanical renewal process?
A2: High variability after mechanical polishing often stems from three common pitfalls:
- Insufficient Cleaning: Residual polishing abrasives (e.g., alumina particles) can remain on the surface, blocking active sites and interfering with electron transfer. Always include a rigorous ultrasonic cleaning step in pure water after polishing [52].
- Inconsistent Protocol: Varying the polishing pressure, duration, or pattern between experiments leads to different surface morphologies and active site densities. Adhere to a strict, documented procedure (e.g., figure-8 pattern for a set time) [52].
- Material Incompatibility: The polishing grit sequence must be appropriate for the electrode hardness. Aggressive polishing on a soft material like carbon paste can irreversibly damage the surface [55].
Q3: When attempting electrochemical renewal, how can I determine the right potential parameters for my specific electrode substrate?
A3: Defining the correct parameters requires a systematic approach:
- Identify the Window: First, establish the electrochemical stability window of your electrode material in your specific electrolyte using cyclic voltammetry. This defines the safe potentials you can apply without degrading the electrode itself [55] [54].
- Target the Contaminant: The renewal protocol must be tailored to the expected contaminants. For organic adsorbates, you may need to cycle to positive potentials to oxidize them. For metal impurities or oxides, a reduction step at negative potentials might be required.
- Validate Effectiveness: The success of electrochemical renewal must be confirmed by a subsequent experiment, such as measuring a standard redox couple (e.g., Ferrocene/Ferrocenium) and confirming that the peak separation and current match expected values for a clean, reversible system [54].
Troubleshooting Common Electrode Renewal Issues
| Problem Description | Likely Cause | Recommended Solution |
|---|---|---|
| High Background Current | Residual surface contaminants or inadequate cleaning after mechanical polish [52] [54]. | Implement a more thorough ultrasonic cleaning protocol. Verify electrolyte purity. Perform a controlled electrochemical renewal cycle after polishing. |
| Poor Reproducibility of Signal | Inconsistent surface renewal between trials; unstable reference electrode [54]. | Standardize renewal protocol (time, pressure, potential sequence). Check reference electrode integrity and ensure proper Luggin capillary placement to minimize iR drop. |
| Visible Surface Scratches | Using an inappropriate polishing grit sequence; skipping finer grit steps [52]. | Follow a progressive polishing regimen (e.g., from 5 μm → 0.3 μm → 0.05 μm alumina). Use a dedicated, clean polishing cloth for each grit size. |
| Drifting Baseline Potential | Unstable electrical contact; formation of a passivating layer on the electrode post-renewal [56] [54]. | Check all electrical connections. For some materials, a brief "conditioning" period at a fixed potential in the electrolyte is needed to stabilize the interface before measurement [56]. |
Detailed Method for Aggressive Mechanical Renewal of Solid Electrodes
This protocol is adapted from standard practices for rejuvenating heavily contaminated electrodes [52].
- Preparation: Affix a piece of 600-grit silicon carbide paper to a stiff, flat glass plate. Add a small volume of deionized water to lubricate.
- Coarse Polishing: Hold the electrode perpendicular to the paper. Using a figure-8 motion while gently rotating the electrode, polish for 2-5 minutes. This step removes gross damage and levels the surface.
- Rinse: Thoroughly rinse the electrode surface with a stream of distilled water to remove all SiC particles.
- Ultrasonic Clean: Suspend the electrode tip in an ultrasonication bath filled with distilled water for 5 minutes to dislodge embedded particles.
- Progress to Finer Grits: Repeat the polishing and cleaning sequence, first with 5 μm alumina on a Nylon pad, then with 0.3 μm alumina on a microcloth, and finally with 0.05 μm alumina on a fresh microcloth. Use a separate pad for each grit.
- Final Clean: After the 0.05 μm step, perform a final thorough rinse and ultrasonic clean. The electrode surface should be mirror-smooth.
Note: This aggressive process removes significant material (250-500 μm) and shortens the electrode's lifespan. It should be reserved for severely contaminated or damaged surfaces [52].
Protocol for In-Situ Mechanical Renewal of Graphite Electrodes
This specialized technique ensures a perfectly standardized surface for each experiment, crucial for reproducibility studies [53].
- Cell Setup: Use a custom electrochemical cell equipped with a cutting tool (e.g., made of artificial sapphire) integrated into the lid.
- Immersion and Polarization: Assemble the cell with the graphite composite electrode, electrolyte, and counter/reference electrodes.
- Surface Renewal: Without breaking the polarization circuit, activate the mechanism to cut off a thin surface layer (~10 μm) of the graphite electrode directly in the electrolyte solution.
- Immediate Measurement: Begin the electrochemical experiment (e.g., CVA or impedance) immediately after renewal. This guarantees that every measurement starts from an identical, freshly exposed surface, free of air-formed layers or previous contamination [53].
Table 1. Comparison of Electrode Surface Renewal Methods Across Different Substrates
| Electrode Substrate | Mechanical Renewal | Electrochemical Renewal | Key Performance Metrics & Notes |
|---|---|---|---|
| Glassy Carbon | Highly Effective. Progressive polishing with alumina slurry is standard [55] [52]. | Conditionally Effective. Can desorb some organics; may not remove all impurities [54]. | Stability: Excellent after mechanical polish. Reproducibility: High with strict protocol. Best for: General purpose use where a pristine, oxide-free surface is needed. |
| Platinum (Pt) | Standard Method. Polishing required to restore active surface area [52]. | Very Effective. Potential cycling in clean acid (e.g., H₂SO₄) reliably reduces oxides and desorbs contaminants [54]. | Stability: Forms oxide layer at positive potentials. Reproducibility: Excellent with electrochemical renewal. Best for: Electrocatalysis studies; easily renewed in situ. |
| Gold (Au) | Standard Method. Similar to Pt [52]. | Limited Usefulness. Surface oxidizes at modest positive potentials, complicating renewal [55]. | Stability: Limited anodic window due to oxidation. Reproducibility: Good with mechanical polishing. Best for: Studies at negative potentials or SAM formation. |
| Graphite | Effective but Variable. Standard polishing works. In-situ cutting provides highest reproducibility [53]. | Less Studied. The semiconductor properties of graphite can complicate the process [53]. | Stability: Good in aprotic solvents (e.g., Propylene Carbonate) [53]. Reproducibility: Excellent with in-situ mechanical cutting [53]. Best for: Battery and capacitor research. |
| Carbon Paste | Ineffective / Damaging. Polishing physically disrupts the soft paste. | Not Applicable. | Renewal Method: Surface is renewed by simply pushing out a small amount of paste and smoothing the surface. Best for: Disposable, single-use surfaces [55]. |
| Screen-Printed Electrodes | Not Possible. | Not Possible. | Renewal Method: These are inherently single-use, disposable devices [56]. |
The Scientist's Toolkit
Table 2. Essential Research Reagent Solutions for Electrode Renewal
| Item | Function & Application |
|---|---|
| Alumina Slurry Suspensions (5 μm, 0.3 μm, 0.05 μm) | A series of abrasive powders in aqueous suspension for progressive mechanical polishing of solid electrodes (Pt, Au, GC) to a mirror finish [52]. |
| Microfiber & Nylon Polishing Cloths | Adhesive-backed cloths used on a flat surface (glass) to hold the alumina slurry during polishing. Nylon pads are for coarser grits, microcloth for finer ones [52]. |
| High-Purity Electrolyte Salts & Solvents | Essential for both electrochemical renewal and subsequent testing. Impurities at ppb levels can poison the electrode surface, invalidating any renewal process [54]. |
| Ultrasonic Cleaning Bath | A critical device used with high-purity water (or solvent) to remove residual alumina particles from the electrode surface after mechanical polishing [52]. |
| Standard Redox Probes (e.g., 1 mM Potassium Ferricyanide) | A solution of a well-characterized, reversible redox couple used to validate the success and reproducibility of a surface renewal protocol by measuring peak separation (ΔEp) and current [54]. |
Experimental Workflow Visualization
The diagram below outlines the logical decision-making process for selecting and validating an electrode surface renewal method, ensuring research reproducibility.
Electrode Renewal Decision Workflow: This chart guides the selection of a surface renewal method based on electrode substrate and contamination type, with validation as a critical final step.
Experimental Protocols for Accelerated Durability Testing
Accelerated test protocols are essential for predicting the service life and durability of electrochemical cells, enabling researchers to identify failure modes rapidly without conducting tests over the full calendar lifetime [57]. The following methodology provides a framework for accelerated durability testing.
General Accelerated Test Profile
The core accelerated test involves cycling the electrochemical cell between open circuit voltage (OCV) and a predetermined operating current density [57]. This cycling accelerates the local redox environment, mimicking long-term degradation processes in a compressed timeframe.
Key Cycling Parameters:
- Current Density: The applied current density during cycling should be identical to that used during steady-state operation to maintain relevance to real-world conditions [57].
- Cycling Frequency: The rate at which cycles are applied significantly impacts acceleration factors [57].
- Operation Temperature: Elevated temperatures can further accelerate degradation processes [57].
- Total Cycles: Studies have implemented up to 1,320,000 cycles to generate statistically significant degradation data [57].
Implementation and Validation
When implementing this protocol, researchers should:
- Compare results from accelerated measurements against cells operated at constant current density to validate correlation [57].
- Study additional parameters including moisture levels, sintering temperature, and operation time to understand their influence on degradation [57].
- Document the acceleration factor achieved; research has demonstrated acceleration of nearly 10 times compared to standard operation [57].
Troubleshooting Guides & FAQs
Common Experimental Issues and Solutions
Table: Troubleshooting Common Problems in Electrochemical Testing
| Problem Symptom | Potential Causes | Recommended Solutions |
|---|---|---|
| Noisy or erratic LPR data | Hydrocarbon layer on working electrode; Poor electrical contact [58] | Rinse cylinder with solvent like acetone; Check spring-loaded ball plunger on corrosion shaft [58] |
| Unusual cyclic voltammogram or different appearance on repeated cycles | Blocked reference electrode frit; Air bubbles blocking electrical contact [59] | Use reference electrode as quasi-reference to test; Check for bubbles between frit and wire [59] |
| Voltage compliance errors | Quasi-reference electrode touching working electrode; Counter electrode removed from solution [59] | Ensure proper electrode spacing; Verify all electrodes are fully submerged [59] |
| Current compliance errors | Working and counter electrodes touching [59] | Inspect electrode arrangement to prevent short circuits [59] |
| Very small, noisy, but otherwise unchanging current | Working electrode not properly connected to cell [59] | Check working electrode connection to electrochemical cell [59] |
| Large reproducible hysteresis in baseline | Charging currents in electrode [59] | Decrease scan rate; Increase analyte concentration; Use smaller working electrode [59] |
Frequently Asked Questions
Should I reuse cylinder inserts/coupons for multiple LPR experiments? No. Cylinder inserts should be considered one-time use electrodes. LPR experiments intentionally corrode the working electrode surface, altering its area and characteristics. Attempting to repolish a used cylinder makes it impossible to know the precise surface area, which is critical for accurate LPR analysis [58].
What is the proper way to set up a reference electrode in corrosion tests? For LPR tests using brine solutions with high salinity, a Luggin capillary may be unnecessary. In high-temperature tests, Luggin capillaries can become blocked by gas bubbles, causing severe electrochemical error. A stable reference electrode is crucial, and combining reference and counter electrodes in a two-electrode setup is not advisable as it reduces potential stability [58].
How should I handle the counter electrode in experiments with two-phase systems? When conducting tests with both aqueous and non-aqueous/oil phases, if the counter electrode is dipped through the oil phase, an oil film can form and block the aqueous phase interface. If this occurs and causes noise, insert the counter electrode directly into the main electrolyte without using a fritted isolation tube [58].
What should I do if my potentiostat shows unusual voltammograms? Follow a systematic troubleshooting procedure: (1) Test the potentiostat and cables using a resistor instead of an electrochemical cell; (2) Use a test chip if available; (3) Connect the reference electrode cable to the counter electrode to check for reference electrode problems; (4) Replace cables and polish the working electrode [59].
Essential Research Reagents and Materials
Table: Key Research Reagent Solutions for Durability Testing
| Item | Function/Application | Key Considerations |
|---|---|---|
| Working Electrode (Cylinder Insert) | Typically 1018 carbon steel coupon; Serves as test substrate for corrosion studies [58] | Remove factory hydrocarbon layer with acetone; Use once only; Ensure good contact with corrosion shaft [58] |
| Counter Electrode | Rod of graphite or stainless steel (e.g., hastelloy); Completes electrical circuit [58] | If using isolation tube, ensure solution on both sides of frit; Watch for oil film blockage in two-phase systems [58] |
| Reference Electrode | Stable potential reference (Ag/AgCl, calomel, or pseudo-reference) [58] | Check for blocked frits or air bubbles; Avoid Luggin capillary in high-temperature tests [58] |
| Electrolyte Solution | Aqueous brine solution typical for LPR tests [58] | High salinity may eliminate need for Luggin capillary [58] |
| Corrosion Cell | Glass vessel (up to 1L capacity) to hold electrolyte and electrodes [58] | Options include basic, water-jacketed, with drain valve, or combination; Choose based on temperature and cleaning needs [58] |
| Alumina Polishing Compound (0.05 μm) | Refresh working electrode surface [59] | Used to remove absorbed species and restore surface between experiments [59] |
Experimental Workflow Visualization
Systematic Troubleshooting Methodology
Technical Support Center
Frequently Asked Questions (FAQs)
Q1: My carbon fiber microelectrode (CFME) signals for dopamine are diminishing over time. What is the cause, and how can I restore performance?
A: Diminished sensitivity is often caused by surface fouling or passivation, which hinders electron transfer. This is a common challenge that undermines analytical performance [33]. You can restore electrode performance through an electrochemical surface renewal procedure.
- Recommended Action: Perform electrochemical regeneration by applying a constant potential of 1.75 V in deionized water for 26.13 minutes [33].
- Mechanism: This treatment regenerates the electrochemically active surface by modifying it with oxygen-containing functional groups, which are crucial for catecholamine adsorption [33].
Q2: I need higher sensitivity for chronic dopamine monitoring. Should I simply use a larger diameter carbon fiber?
A: While increasing fiber diameter can improve mechanical robustness and in vitro sensitivity, it can cause significant tissue damage and reduced in vivo performance. A study showed that 30 µm bare CFMEs had 2.7-fold higher in vitro sensitivity than 7 µm CFMEs, but their in vivo dopamine signals were significantly lower, likely due to insertion trauma [60].
- Recommended Solution: Consider using a 30 µm cone-shaped CFME. This design combines the strength of a larger fiber with a tapered tip, which was shown to produce a 3.7-fold improvement in in vivo dopamine signals compared to bare 30 µm CFMEs and significantly reduce glial activation [60].
Q3: What is a simple pre-treatment to enhance the sensitivity of my CFME for a range of neurotransmitters?
A: Electrochemical treatment in potassium hydroxide (KOH) is a fast and effective method to increase sensitivity.
- Protocol for Standard CFMEs: Apply 1.5 V vs. Ag/AgCl for 3 minutes while the electrode is dipped in 1 M KOH [61]. This etches the surface and adds oxygen functional groups, improving sensitivity to dopamine, epinephrine, norepinephrine, and serotonin by roughly 2-fold [61].
Troubleshooting Guides
Problem: Inconsistent or Drifting Baseline Current During FSCV
| Potential Cause | Explanation | Solution |
|---|---|---|
| Un-stabilized Electrode | A new or regenerated electrode requires time for the background current to stabilize in the buffer solution. | After fabrication or any pre-treatment, stabilize the electrode in PBS buffer by applying the FSCV waveform until the background current is constant (typically 5-10 minutes) [61]. |
| Surface Contamination | Adsorbed contaminants from the biological environment or storage can foul the surface. | Implement the deionized water regeneration protocol [33] or use the KOH pre-treatment before experiments to clean and activate the surface [61]. |
| Mechanical Erosion | The carbon fiber surface degrades over time with repeated use, especially in vivo. | Benchmark against known standards. Note that cone-shaped 30 µm CFMEs showed a 4.7-fold increase in lifespan in erosion tests compared to standard 7 µm CFMEs [60]. |
Problem: Low Signal-to-Noise Ratio in Dopamine Detection
| Potential Cause | Explanation | Solution |
|---|---|---|
| Sub-optimal Electrode Geometry | A standard cylindrical fiber may not offer the best combination of sensitivity and biocompatibility. | Switch to a cone-shaped geometry, which minimizes tissue compression during insertion, improving the quality of the in vivo signal [60]. |
| Insufficient Surface Activation | The electrode surface lacks sufficient defect sites and oxygen functional groups that enhance sensitivity. | Apply the KOH electrochemical treatment to etch the surface and add oxygen functional groups, which can double the dopamine response [61]. |
| Incorrect Waveform Parameters | The FSCV waveform may not be optimized for the renewed electrode surface. | Use a standard dopamine waveform (e.g., -0.4 V to 1.3 V at 400 V/s, 10 Hz) and ensure the pre-conditioning waveform (e.g., -0.4 V to 1.5 V) is applied [60]. |
Experimental Data & Protocols
Quantitative Performance Benchmarking of CFME Designs
The following table summarizes key performance metrics from recent studies for different CFME configurations, providing a benchmark for evaluating your own renewed electrodes.
Table 1: In Vitro and In Vivo Performance of Various CFME Designs for Dopamine Detection
| CFME Design | In Vitro Sensitivity (pA/µm²) | In Vivo Dopamine Signal (nA) | Key Characteristics | Source |
|---|---|---|---|---|
| 7 µm Standard CFME | 12.2 ± 4.9 | 24.6 ± 8.5 | Standard design; minimal initial tissue damage | [60] |
| 30 µm Bare CFME | 33.3 ± 5.9 | 12.1 ± 8.1 | High mechanical strength & in vitro sensitivity; causes tissue damage | [60] |
| 30 µm Cone-Shaped CFME | Data not specified | 47.5 ± 19.8 | Mitigates insertion damage; enhances in vivo signal & longevity | [60] |
| KOH-treated CFME | ~2-fold increase reported | Not specified | Improved sensitivity for multiple cationic neurotransmitters | [61] |
| DI Water Regenerated CFME | LOD of 31 nM for DA | Not specified | Restores performance of fouled electrodes; useful for complex bio-environments | [33] |
Detailed Experimental Protocol: KOH Electrochemical Treatment
This protocol is adapted from a 2023 study that investigated the treatment for multiple neurotransmitters [61].
- Solution Preparation: Prepare a 1 M KOH solution as the treatment solution.
- Electrode Setup: Use a standard three-electrode system with the CFME as the working electrode and an Ag/AgCl reference electrode.
- Application of Potential: Apply a constant potential of 1.5 V vs. Ag/AgCl to the CFME while it is dipped in the 1 M KOH solution.
- Treatment Duration: Maintain this potential for 3 minutes.
- Post-Treatment Rinse: Rinse the electrode thoroughly with deionized water.
- Stabilization in Buffer: Before detection, place the treated CFME in PBS buffer (pH 7.4) and run the FSCV waveform until the background current stabilizes (5-10 minutes).
Workflow for CFME Renewal and Benchmarking
The following diagram outlines the logical workflow for evaluating and implementing CFME renewal strategies within a research context focused on reproducibility.
The Scientist's Toolkit
Table 2: Essential Research Reagent Solutions for CFME Renewal and Dopamine Detection
| Item | Function / Explanation | Example Specification / Notes |
|---|---|---|
| Potassium Hydroxide (KOH) | Electrochemical pre-treatment solution. Etches the carbon surface and introduces oxygen-containing functional groups, boosting sensitivity [61]. | 1 M solution; apply 1.5 V for 3 minutes [61]. |
| Deionized (DI) Water | Electrolyte-free medium for electrode regeneration. A simple method to restore the electrochemical performance of inactivated or fouled CFMEs [33]. | Apply 1.75 V for 26.13 minutes for regeneration [33]. |
| Tris Buffer | A stable buffer for in vitro electrochemical testing and calibration. Provides electrochemical stability and signal consistency in controlled conditions [60]. | pH 7.4, with ionic components like NaCl, KCl, CaCl₂ [60]. |
| Phosphate Buffered Saline (PBS) | A biological buffer that mimics ionic strength of physiological fluids. Used for stabilizing electrodes and in vitro testing post-renewal [61]. | pH 7.4; used for final electrode stabilization and calibration [61]. |
| Dopamine Stock Solution | The primary analyte for calibration and benchmarking. | 1-10 mM stock in 0.1 M HClO₄ to prevent oxidation; dilute in buffer for working concentrations [61]. |
| Carbon Nanotube Yarn (CNTY) | Alternative nanomaterial for microelectrodes. Can be similarly treated with KOH for enhanced sensitivity, offering another platform for renewal studies [61]. | 50 µm diameter; KOH treatment time is shorter (1 min) to avoid cracking [61]. |
Conclusion
Achieving high reproducibility in solid electrode surface renewal hinges on a deep understanding of the underlying mechanisms and a disciplined, method-specific approach. The convergence of electrochemical and mechanical strategies offers a powerful toolkit, but its success is measured by rigorous validation using standardized metrics for sensitivity, stability, and limit of detection. Future progress depends on developing universally accepted protocols and exploring novel 'self-renewing' materials. For biomedical research, these advances promise to unlock more reliable in vivo monitoring, robust point-of-care diagnostics, and accelerated drug discovery by ensuring that electrochemical data is built upon a foundation of consistent and regenerable sensor interfaces.
References
- 1. Passivation (chemistry) [https://en.wikipedia.org/wiki/Passivation_(chemistry)]
- 2. Passivation - an overview | ScienceDirect Topics [https://www.sciencedirect.com/topics/engineering/passivation]
- 3. Electrode Passivation → Area [https://energy.sustainability-directory.com/area/electrode-passivation/]
- 4. Renewable Solid Electrodes in Microfluidics [https://pubmed.ncbi.nlm.nih.gov/27748597/]
- 5. How to Improve the Performance of Electrochemical ... [https://www.mdpi.com/2227-9040/9/1/12]
- 6. A guide to troubleshooting metal sacrificial anodes for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11041367/]
- 7. In Situ Renewal of Working Surfaces of Solid... [https://www.ovid.com/journals/janc/fulltext/00009689-200560120-00010~in-situ-renewal-of-working-surfaces-of-solid-indicator]
- 8. Spectroscopic investigations of the fouling process on ... [https://www.sciencedirect.com/science/article/abs/pii/S0376738810007878]
- 9. Structure of interfaces on mechanically renewed Sn, Pb ... [https://www.sciencedirect.com/science/article/abs/pii/S1572665721009784]
- 10. Troubleshooting the functionalization of gold screen printed ... [https://www.zimmerpeacock.com/2025/07/13/troubleshooting-the-functionalization-of-gold-screen-printed-electrodes/]
- 11. Oxygen-Containing Functional Groups Regulating the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8438096/]
- 12. Effects of Oxygen-Containing Functional Groups on the ... [https://www.mdpi.com/2079-4991/13/2/262]
- 13. Effects of oxygen-containing functional groups on carbon ... [https://www.sciencedirect.com/science/article/pii/S0264127523003672]
- 14. Dynamic evolution of self-renewal Fe–N–C catalysts for the ... [https://pubs.rsc.org/en/content/articlehtml/2025/ey/d5ey00092k]
- 15. Benchmarking the reproducibility of all-solid-state battery ... [https://www.nature.com/articles/s41560-024-01634-3]
- 16. Unlocking catalytic longevity: a critical review of catalyst ... [https://pubs.rsc.org/en/content/articlehtml/2025/ya/d5ya00015g]
- 17. for Energy and Environmental... Advanced Catalytic Materials [https://pubs.rsc.org/en/journals/articlecollectionlanding?sercode=cy&themeid=4ee006a8-2cb6-490c-9fa5-80ecb2425157]
- 18. Highly reproducible solid contact ion selective electrodes: ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267021001306]
- 19. An innovative DMAIC and response surface methodology ... [https://www.sciencedirect.com/science/article/abs/pii/S0360544225048157]
- 20. Complete Guide to Electroplating Defects & Issues [https://www.sharrettsplating.com/blog/electroplating-defects-issues/]
- 21. A guide to troubleshooting metal sacrificial anodes for organic ... [https://pubs.rsc.org/en/content/articlehtml/2024/sc/d3sc06885d]
- 22. Surface renewal driven copper recovery by cementation in ... [https://www.nature.com/articles/s41598-025-10845-x]
- 23. Is reproducibility inside the bag? Special issue ... [https://www.sciencedirect.com/science/article/pii/S1350417717301372]
- 24. How to Diagnose Surface Pattern Problems in Grinding [https://www.cdtusa.net/blog/diagnosing-surface-pattern-problems]
- 25. Application of the surface renewal technique in two types ... [https://www.sciencedirect.com/science/article/abs/pii/S0168192315000118]
- 26. Surface Renewal: An Advanced Micrometeorological ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4048044/]
- 27. New trends in the electrochemical sensing of dopamine - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3608872/]
- 28. Unveiling the PEDOT-polypyrrole hybrid electrode for ... [https://www.nature.com/articles/s41598-024-82355-1]
- 29. Dopamine sensing and energy storage application [https://www.sciencedirect.com/science/article/abs/pii/S1387700324015429]
- 30. Highly Sensitive Electrochemical Detection of Dopamine ... [https://www.sciencedirect.com/science/article/pii/S1452398125002676]
- 31. Detection of Dopamine Using Hybrid Materials Based on ... [https://www.mdpi.com/2073-4344/15/2/116]
- 32. Carbon nanotubes-based electrode materials for ... [https://www.sciencedirect.com/science/article/abs/pii/S0026265X25007076]
- 33. Electrochemical surface modification and regeneration of ... [https://www.sciencedirect.com/science/article/pii/S1872204025000416]
- 34. Basic Principles [https://www.basinc.com/manuals/LC_epsilon/Principles/Basic/basic]
- 35. Catalyst Deactivation - an overview [https://www.sciencedirect.com/topics/chemical-engineering/catalyst-deactivation]
- 36. Catalyst Degradation Mechanisms → Term [https://energy.sustainability-directory.com/term/catalyst-degradation-mechanisms/]
- 37. Three Sources of Catalyst Deactivation and How To ... [https://www.chemcatbio.org/resources/technology-briefs/2023-tech-brief-three-sources-of-catalyst-deactivation]
- 38. A method for in situ auto-renewal of the surface of glassy ... [https://www.sciencedirect.com/science/article/abs/pii/S0022072803003954]
- 39. Troubleshooting and Optimization Tips for SPR Experiments [https://www.creative-proteomics.com/resource/troubleshooting-optimization-tips-for-spr-experiments.htm]
- 40. Strategies for Biofouling Control: A Review from an ... [https://www.mdpi.com/2079-6412/15/10/1185]
- 41. Experimental characterization of adsorbed protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4312349/]
- 42. Biofouling prevention and control technology for marine ... [https://www.sciencedirect.com/science/article/pii/S0025326X25014006]
- 43. Developments and Ongoing Challenges for Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8522203/]
- 44. Experimental studies on the desorption of adsorbed ... [https://www.sciencedirect.com/science/article/abs/pii/S0268005X04001626]
- 45. Topology optimization of porous electrodes for ... [https://www.sciencedirect.com/science/article/pii/S1385894725026415]
- 46. Towards the development of an integrated, user friendly ... [https://www.sciencedirect.com/science/article/abs/pii/S0263224125031355]
- 47. A Reliable and Simple Voltammetric Method for Analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12567795/]
- 48. Working Electrodes for Electrochemistry | Sizes, Materials, ... [https://www.sciencegears.com.au/working-electrode]
- 49. Influence of Surface Treatments on the Electrochemical ... [https://www.mdpi.com/2673-4532/6/1/9]
- 50. Electrochemical hydrogen storage: Critical parameters and ... [https://www.sciencedirect.com/science/article/abs/pii/S0378775325023171]
- 51. Cerium-doped iron oxide nanostructures used as a ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr03310a]
- 52. Electrode Polishing Guide [https://pineresearch.com/support-article/electrode-polishing-guide/]
- 53. Specific features of the interaction of a mechanically ... [https://www.sciencedirect.com/science/article/abs/pii/S157266572030357X]
- 54. Error, reproducibility and uncertainty in experiments for ... [https://www.nature.com/articles/s41467-022-34594-x]
- 55. C. Working Electrodes [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Supplemental_Modules_(Analytical_Chemistry)/Analytical_Sciences_Digital_Library/Courseware/Analytical_Electrochemistry%3A_The_Basic_Concepts/05_Experimental_Hardware/C._Working_Electrodes]
- 56. Stability and reproducibility study for the development of ... [https://www.sciencedirect.com/science/article/pii/S2666831925001572]
- 57. Accelerated test protocols to predict service life and ... [https://www.sciencedirect.com/science/article/pii/S2949821X22000011]
- 58. Troubleshooting LPR Experiments [https://pineresearch.com/support-article/troubleshooting-lpr-experiments/]
- 59. Troubleshooting Cyclic Voltammetry and Voltammograms [https://www.ossila.com/pages/cyclic-voltammetry-troubleshooting]
- 60. Frontiers | Improved longevity and in vivo performance of... [https://www.frontiersin.org/journals/bioengineering-and-biotechnology/articles/10.3389/fbioe.2025.1579380/full]
- 61. Electrochemical treatment in KOH improves carbon nanomaterial... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10788926/]